

Single-cell RNA sequencing (scRNA-seq) has revolutionized our understanding of cellular diversity and function. However, the presence of batch effects in scRNA-seq data complicates comparative analyses across different samples, individuals, and conditions. While batch correction methods exist, they often lead to overcorrection and the loss of important biological variability.
In a recent study, researchers at the Ludwig Institute for Cancer Research introduce STACAS, a novel batch correction method for scRNA-seq data. Unlike traditional approaches, STACAS leverages prior knowledge of cell types to preserve biological variability during data integration. By incorporating information about cell types, STACAS ensures that relevant biological signals are retained while correcting for batch effects.
Schematic of semi-supervised STACAS integration method
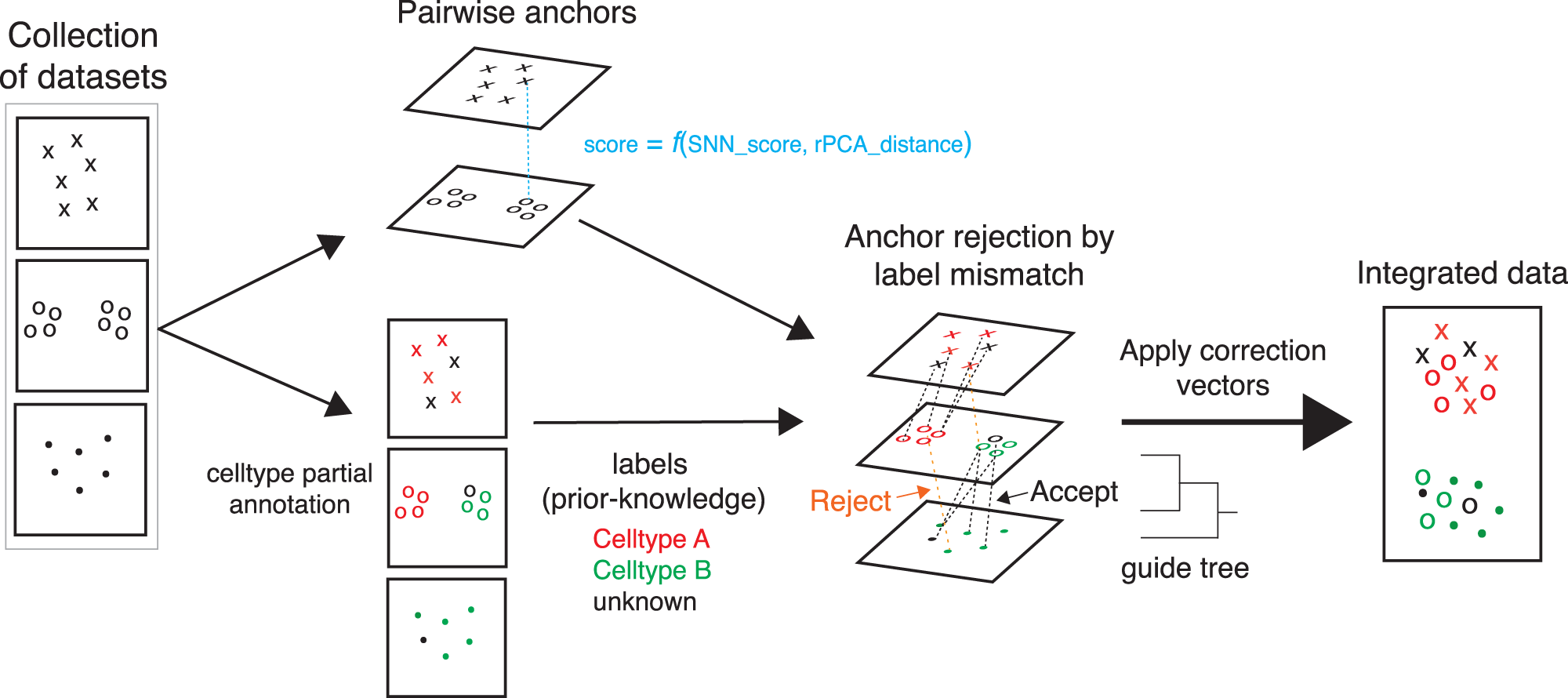
The algorithm identifies integration anchors between all pairs of datasets from a shared nearest neighbors (SNN) graph. These are expected to be cells of the same type across batches and are used to calculate batch effects. Integration anchors are weighted by a score that combines a SNN anchor consistency score (based on the overlap of shared neighbors) and a score based on rPCA distance (how similar are cells of one dataset to the corresponding anchor cells in a second dataset projected into the PCA space of the latter). If cell type labels are available, they can be provided as input to the algorithm. When cell type labels between two cells of an anchor are inconsistent, the anchor is rejected with a predefined probability and in that case will not contribute to batch effect correction. Finally, the sum of retained, weighted integration anchor scores is used to calculate global similarities between datasets and to derive a guide tree that will determine the order in which the datasets are to be integrated.
Through an open-source benchmark, the researchers demonstrate that STACAS outperforms existing unsupervised methods and even supervised approaches like scANVI and scGen. Notably, STACAS is capable of handling large datasets and remains robust even when input cell type labels are incomplete or imprecise, which is common in real-life integration tasks.
The key strength of STACAS lies in its semi-supervised approach, where it utilizes prior cell type information to guide batch correction. This ensures that the integration process maintains biological variability, allowing for more accurate downstream analyses.
The implications of STACAS are significant for the field of single-cell genomics. By providing a flexible framework for semi-supervised batch effect correction, STACAS enables researchers to extract meaningful insights from scRNA-seq data with greater confidence. This approach not only enhances the reliability of comparative analyses but also facilitates the discovery of novel biological insights.
In conclusion, STACAS represents a promising advancement in the field of single-cell RNA sequencing. Its ability to preserve biological variability while effectively correcting batch effects underscores its importance in unraveling the complexities of cellular function and diversity. As single-cell genomics continues to evolve, methods like STACAS will play a crucial role in advancing our understanding of the intricacies of cellular biology.
Availability – STACAS is available as a R package at https://github.com/carmonalab/STACAS and https://doi.org/10.5281/zenodo.10402054
Andreatta M, Hérault L, Gueguen P, Gfeller D, Berenstein AJ, Carmona SJ. (2024) Semi-supervised integration of single-cell transcriptomics data. Nat Commun 15(1):872. [article]


