

Understanding the molecular activity of different cell types is essential for unraveling the mysteries of life. Transcriptomic data, which provides insights into the genes expressed by cells, serves as a valuable tool in this pursuit. However, analyzing RNA sequencing (RNA-seq) data can sometimes be challenging due to technical limitations, such as insufficient read depth or the phenomenon known as gene dropout. Additionally, RNA-seq experiments may detect lowly expressed mRNAs that are not necessarily biologically relevant but rather products of leaky transcription.
To address these challenges and accurately represent a cell type’s functional transcriptome, researchers at the University College London have proposed a novel approach. By compiling numerous bulk RNA-seq datasets into a compendium and applying established classification models, they aim to distinguish between transcripts resulting from active gene expression and those from leaky transcription.
The BulkECexplorer compendium, short for bulk RNA-seq endothelial cell explorer, comprises a collection of 240 bulk RNA-seq datasets from five distinct vascular endothelial cell subtypes. This resource serves as a treasure trove of transcriptomic information, providing researchers with valuable insights into the gene expression profiles of endothelial cells. By analyzing these datasets collectively, researchers can gain a deeper understanding of the molecular activity within these cell types.
BulkECexplorer online app display
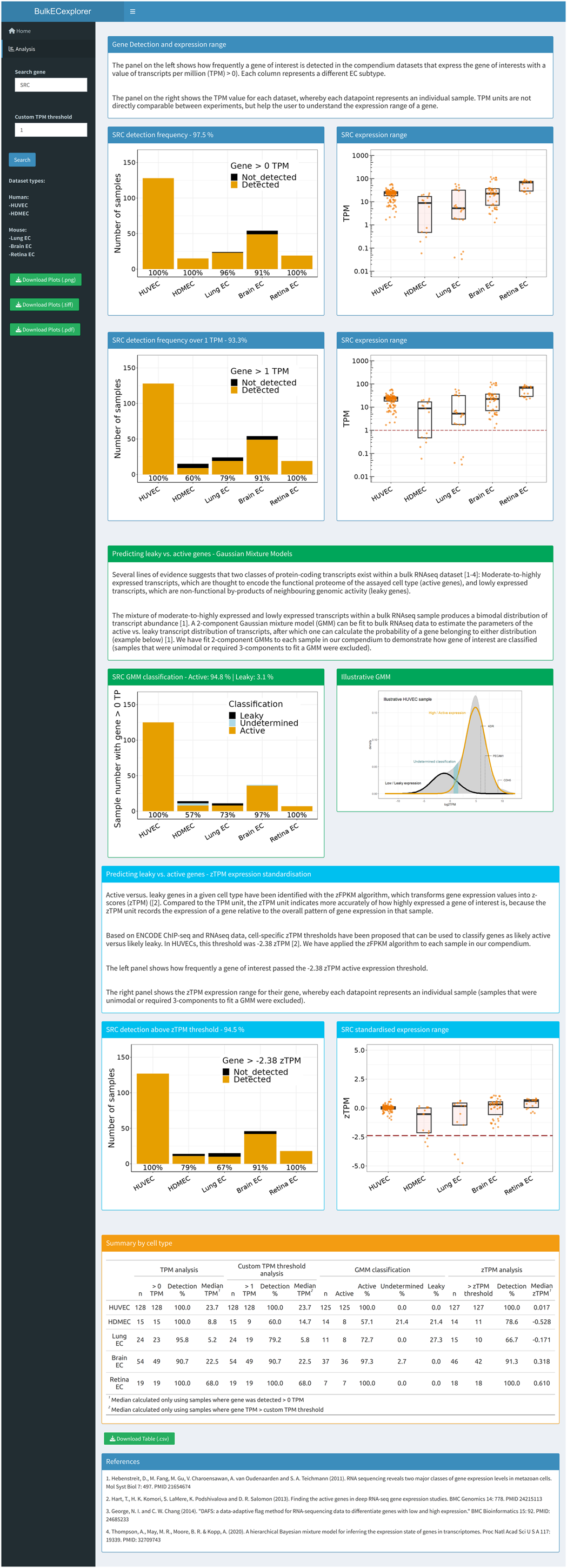
The image shows a snapshot of the output of the BulkECexplorer when queried for a gene of interest (for example, SRC). The blue section displays the gene detection rate and expression range. Top left box, stacked bar chart depicting the number of datasets with SRC >0 TPM, resolved by EC subtype. The percentage of datasets with SRC >0 TPM in each EC subtype is reported below each bar. The percentage of datasets with SRC >0 TPM across all datasets, independently of subtype, is reported above the bar graph. Top right box, boxplots of SRC TPM values for individual datasets, resolved by EC subtype, including the median (center line). The bottom boxes show the corresponding data with a default ‘>1 TPM’ expression threshold that can be customized. The red dashed line (bottom right box) indicates the 1 TPM gene expression threshold. The green section summarizes data obtained by predicting leaky versus active genes using GMMs. Left box, stacked bar chart depicting the number of datasets in which SRC expression was classified as active, leaky or undetermined, resolved by EC subtype. The percentage of datasets in which SRC expression was classified as active in each EC subtype is reported below each bar. The percentage of datasets in which SRC expression was classified as active versus leaky across all datasets is reported above the bar chart. Right box, GMM for a representative HUVEC dataset; expression values for three core EC genes are indicated. The cyan section summarizes data obtained by predicting leaky versus active genes using zTPM expression standardization for each dataset. Left box, stacked bar chart depicting the number of datasets in which SRC expression was above the −2.38 zTPM threshold, resolved by EC subtype. The percentage of datasets in which SRC was detected above the threshold in each EC subtype is reported below each bar. The percentage of datasets in which SRC was detected above the threshold across all datasets is reported above the bar chart. Right box, boxplots of SRC zTPM values for individual datasets, resolved by EC subtype, including the median (center line). The red dashed line indicates the −2.38 zTPM gene expression threshold. The orange section provides a summary by cell type for the number of datasets analyzed per EC subtype alongside outputs for the analysis of TPM values and the GMM versus zTPM predictions.
One of the key features of the BulkECexplorer compendium is its ability to predict whether detected transcripts are likely products of active gene expression or leaky transcription. This distinction is crucial for accurately interpreting transcriptomic data and identifying genes that play functional roles within endothelial cells. By leveraging established classification models, researchers can navigate through the vast transcriptomic landscape with greater confidence, distinguishing signal from noise and uncovering novel insights into endothelial cell biology.
Beyond its immediate applications in vascular biology research, the BulkECexplorer compendium holds promise as a blueprint for developing similar tools for other cell types. By compiling and analyzing bulk RNA-seq datasets from diverse cell populations, researchers can gain a comprehensive understanding of cellular transcriptomes across various tissues and organisms. This holistic approach to transcriptomic analysis opens new avenues for research and discovery in fields ranging from developmental biology to disease pathogenesis.
The BulkECexplorer compendium represents a significant advancement in the field of transcriptomics, offering researchers a powerful tool for exploring the functional transcriptome of vascular endothelial cells. By harnessing the collective power of bulk RNA-seq data and classification models, researchers can uncover hidden patterns within transcriptomic datasets and gain deeper insights into cellular biology. As we continue to unravel the complexities of gene expression, compendiums like BulkECexplorer pave the way for a deeper understanding of the molecular mechanisms underlying life itself.
Availability – Code is available at https://github.com/ruhrberg.
Brash JT, Diez-Pinel G, Colletto C et al. (2024) The BulkECexplorer compiles endothelial bulk transcriptomes to predict functional versus leaky transcription. Nat Cardiovasc Res [Epub ahead of print]. [article]


