

Individual cells can assume a variety of molecular and phenotypic states and recent studies indicate that cells can rapidly adapt in response to therapeutic stress. Such phenotypic plasticity may confer resistance, but also presents opportunities to identify molecular programs that could be targeted for therapeutic benefit. Approaches to quantify tumor-drug responses typically focus on snapshot, population-level measurements. While informative, these methods lack lineage and temporal information, which are particularly critical for understanding dynamic processes such as cell state switching. As new technologies have become available to measure lineage relationships, modeling approaches will be needed to identify the forms of cell-to-cell heterogeneity present in these data.
Researchers at UCLA applied a lineage tree-based adaptation of a hidden Markov model that employs single cell lineages as input to learn the characteristic patterns of phenotypic heterogeneity and state transitions. In benchmarking studies, the researchers demonstrated that the model successfully classifies cells within experimentally-tractable dataset sizes. As an application, they analyzed experimental measurements in cancer and non-cancer cell populations under various treatments. They found evidence of multiple phenotypically distinct states, with considerable heterogeneity and unique drug responses. In total, this framework allows for the flexible modeling of single cell heterogeneity across lineages to quantify, understand, and control cell state switching.
The tHMM model
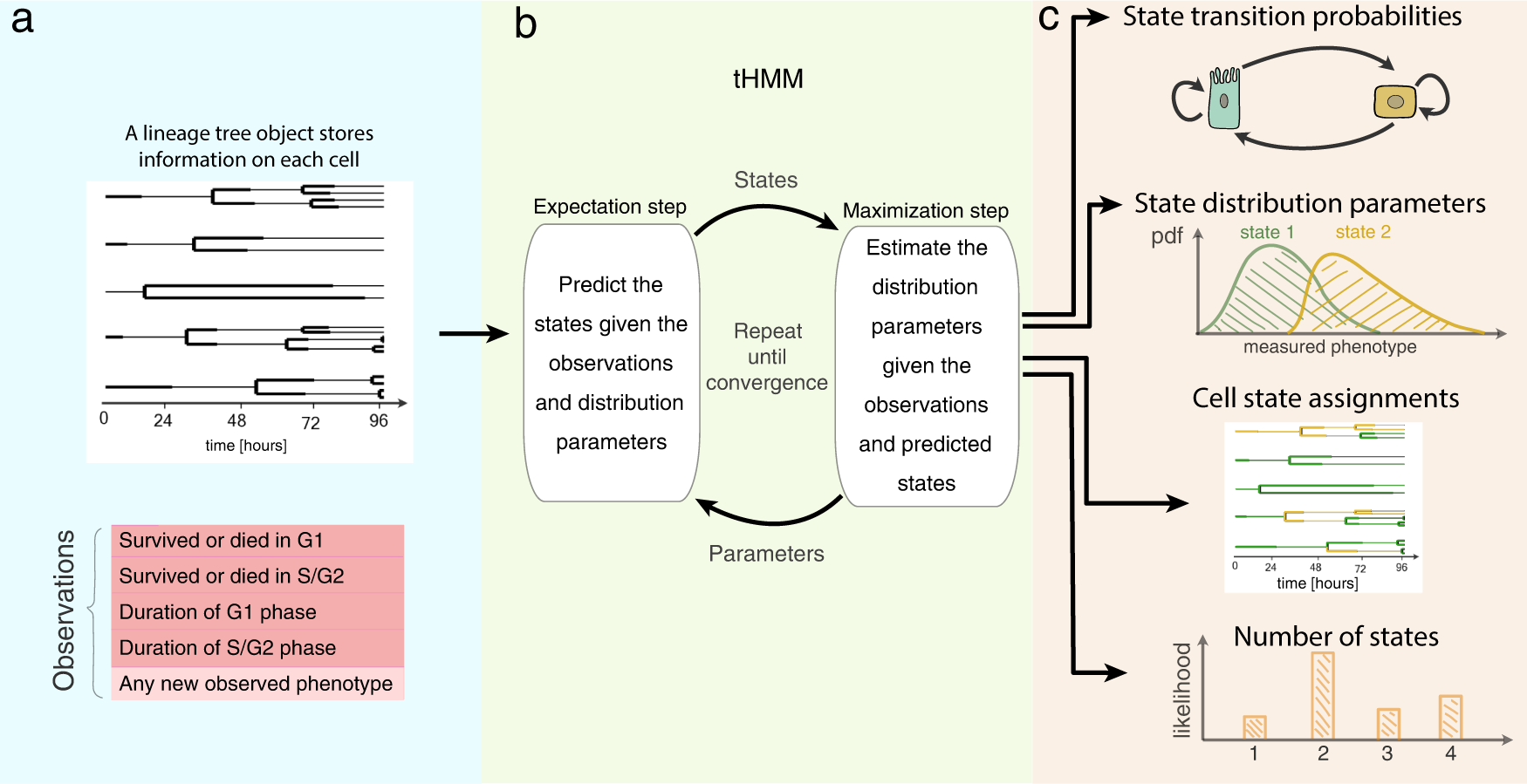
a Input data takes the form of single-cell measurements across time, where the lineage relationship between cells is known. b The fitting process includes expectation and maximization steps, where model parameters are iteratively updated until convergence. c Output predictions of the model after fitting including the tree of inferred cell states, probabilities of transition between each state, starting abundance of each cell state, and distributions that describe the behavior of cells within each state. The model likelihood can be used to estimate the number of distinguishable cell states.
Availability – All analysis were implemented in Python v3.9 and can be found at https://github.com/meyer-lab/tHMM.
Mohammadi F, Visagan S, Gross SM, Karginov L, Lagarde JC, Heiser LM, Meyer AS. (2022) A lineage tree-based hidden Markov model quantifies cellular heterogeneity and plasticity. Commun Biol 5(1):1258. [article]


