

The novel terminator-assisted solid-phase complementary DNA amplification and sequencing (TAS-Seq) method provides high-precision data on gene expression
Single-cell RNA sequencing (scRNA-seq) is one of the most important methods to study biological function in cells, but it is limited by potential inaccuracies in the data it generates. Now, researchers from the Tokyo University of Science have developed a new method called terminator-assisted solid-phase complementary DNA amplification and sequencing (TAS-Seq), which overcomes these limitations and provides higher-precision data than existing scRNA-seq platforms.
The advent of single-cell RNA sequencing (scRNA-seq) has revolutionized the fields of medicine and biology by providing the ability to study the inner workings of thousands of cells at one go. But scRNA-seq methods are limited by potential inaccuracies in determining cell composition and inefficient complementary DNA (cDNA) amplification―a process by which a double-stranded DNA that ‘complements’ the single-stranded RNA is generated and replicated millions of times―by the commonly-used template-switching reaction.
Recently, a research team from Japan, led by Assistant Prof. Shigeyuki Shichino and Prof. Kouji Matsushima of Tokyo University of Science, has developed a new and improved technique for scRNA-seq. The new method, terminator-assisted solid-phase cDNA amplification and sequencing (TAS-Seq), uses simple materials and equipment to provide higher-precision scRNA-seq data than current, widely-used technologies.
“Our technique, TAS-Seq, combines genetic detection sensitivity, robustness of reaction efficiency, and accuracy of cellular composition to enable us to capture important cellular information,” reveals Assistant Prof. Shichino.
The study was published in Communications Biology on June 27, 2022. The research team also included Associate Prof. Satoshi Ueha of Tokyo University of Science, Prof. Taka-aki Sato of the University of Tsukuba, and Prof. Shinichi Hashimoto of Wakayama Medical University.
TAS-Seq uses a template independent enzyme for cDNA amplification called terminal transferase (TdT). But TdT is difficult to handle. To surmount this challenge, the research team included dideoxynucleotide phosphate (ddNTP) as a ‘terminator’ for the cDNA amplification reaction.
“ddNTP spike-in, specifically dideoxycytidine phosphate (ddCTP), stops the excessive extension of polyN-tail by TdT in a stochastic manner, and greatly reduces the technical difficulties of the TdT reaction,” explains Assistant Prof. Shichino.
TAS-Seq also uses a nanowell/bead-based scRNA-seq platform, which allows the isolation of single cells in tissue samples, thereby decreasing cell sampling bias and improving the accuracy of cell composition data.

The research team then verified the efficiency of TAS-Seq and compared it to the current, widely used scRNA-seq techniques, 10X Chromium V2 and Smart-seq2, using murine and human lung tissue samples. They found that TAS-Seq could not only detect more genes overall, but also identify more highly variable genes, when compared to major scRNA-seq platforms.
Assistant Prof. Shichino says, “We found that TAS-Seq may outperform 10X Chromium V2 and Smart-seq2 in terms of gene detection sensitivity and gene drop-out rates, indicating that TAS-Seq might be one of the most sensitive high-throughput scRNA methods. We can detect genes across a wide range of expression levels more uniformly and also detect growth factor and interleukin genes more robustly.”
Principles of TAS-Seq
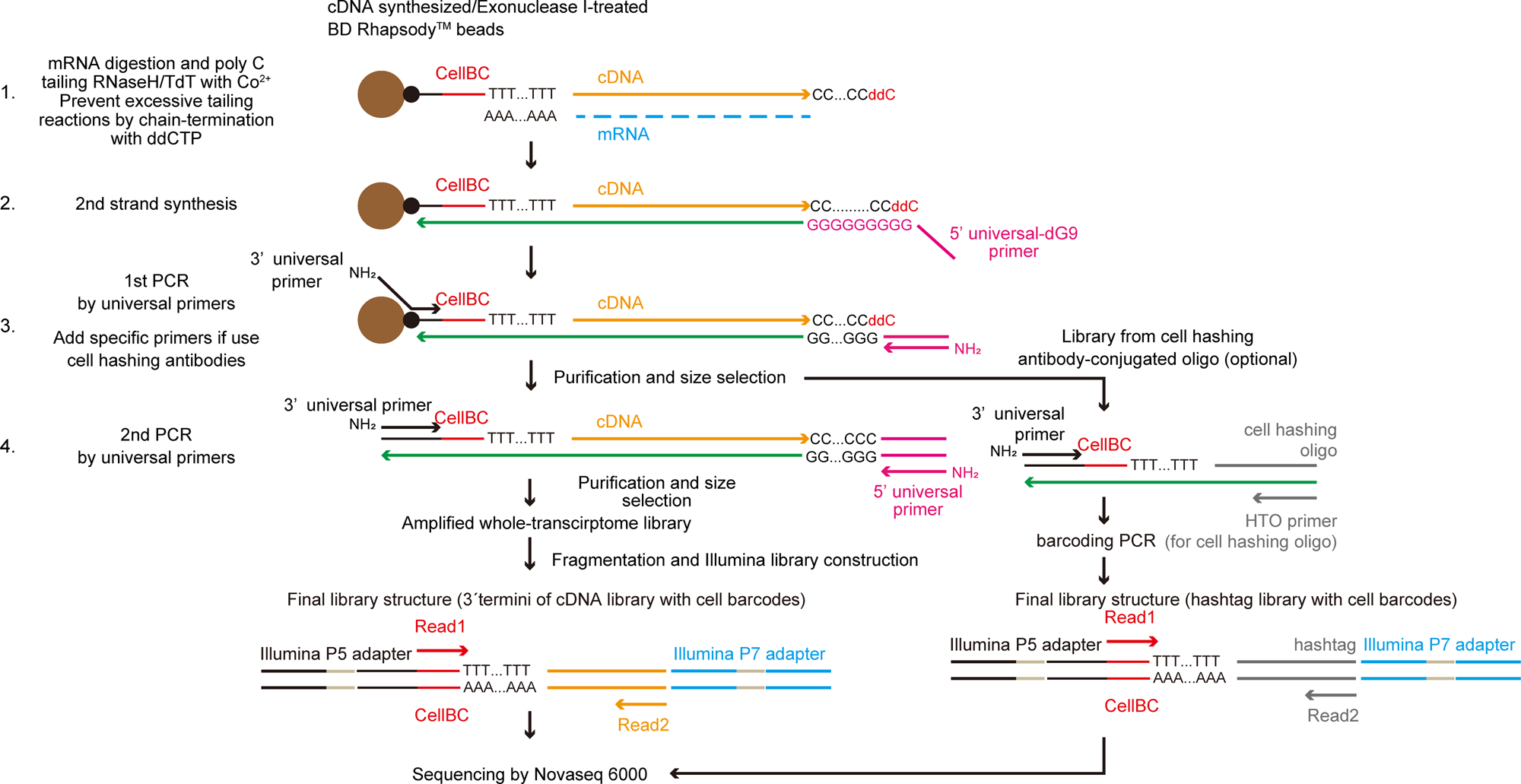
Diagram of the TAS-Seq library preparation workflow. First, cDNA synthesis on BD Rhapsody magenic beads were performed. After Exonuclease I treatment, dC-tailing reaction was performed by TdT/RNase H. Tailing reaction was stochastically stopped by ddCTP incorporation into 3′ termini (1). Next, second-strand synthesis was performed using 5’universal-dG9 primer and PCR master mix (2). Then, 1st PCR was performed by appropriate primers (3′ and 5′ universal primer (if only amplify cDNA) or 3′, 5′ universal primer and HTO primer (if use cell hashing antibodies)) (3). Resultant libraries were size-selected and amplified by 2nd PCR (4). cDNA libraries were fragmented, ligated, and truncated with Illumina P7 adapters, and 3′ termini of cDNA molecules with cell barcodes were enriched by PCR amplification. During the PCR step, we introduced unique-dual barcodes to the Illumina adapters to minimize index hopping. Finally, libraries were sequenced by Illumina Novaseq 6000.
Source – Tokyo University of Science
Shichino S, Ueha S, Hashimoto S et al. (2022) TAS-Seq is a robust and sensitive amplification method for bead-based scRNA-seq. Commun Biol [Epub ahead of print]. [article]


