

In personalized oncology, gene expression profiling has become increasingly vital for tailoring treatment strategies to individual patients. However, ensuring consistent and comparable expression measurements across different RNA sequencing (RNA-seq) protocols is essential for accurate analysis. Addressing this challenge, a recent study by researchers at BostonGene has introduced optimized protocols and a cutting-edge algorithm to enhance the reliability of RNA-seq data obtained from diverse sample preparation methods.
The study focuses on optimizing an exome capture (EC)-based protocol for processing formalin-fixed, paraffin-embedded samples, which are commonly used in cancer diagnosis and research. Additionally, a novel machine-learning algorithm, named Procrustes, has been developed to mitigate batch effects across RNA-seq data generated from various protocols, including EC-based and poly-A RNA-seq methods.
Optimization of exome capture-based (EC) RNA-seq protocol
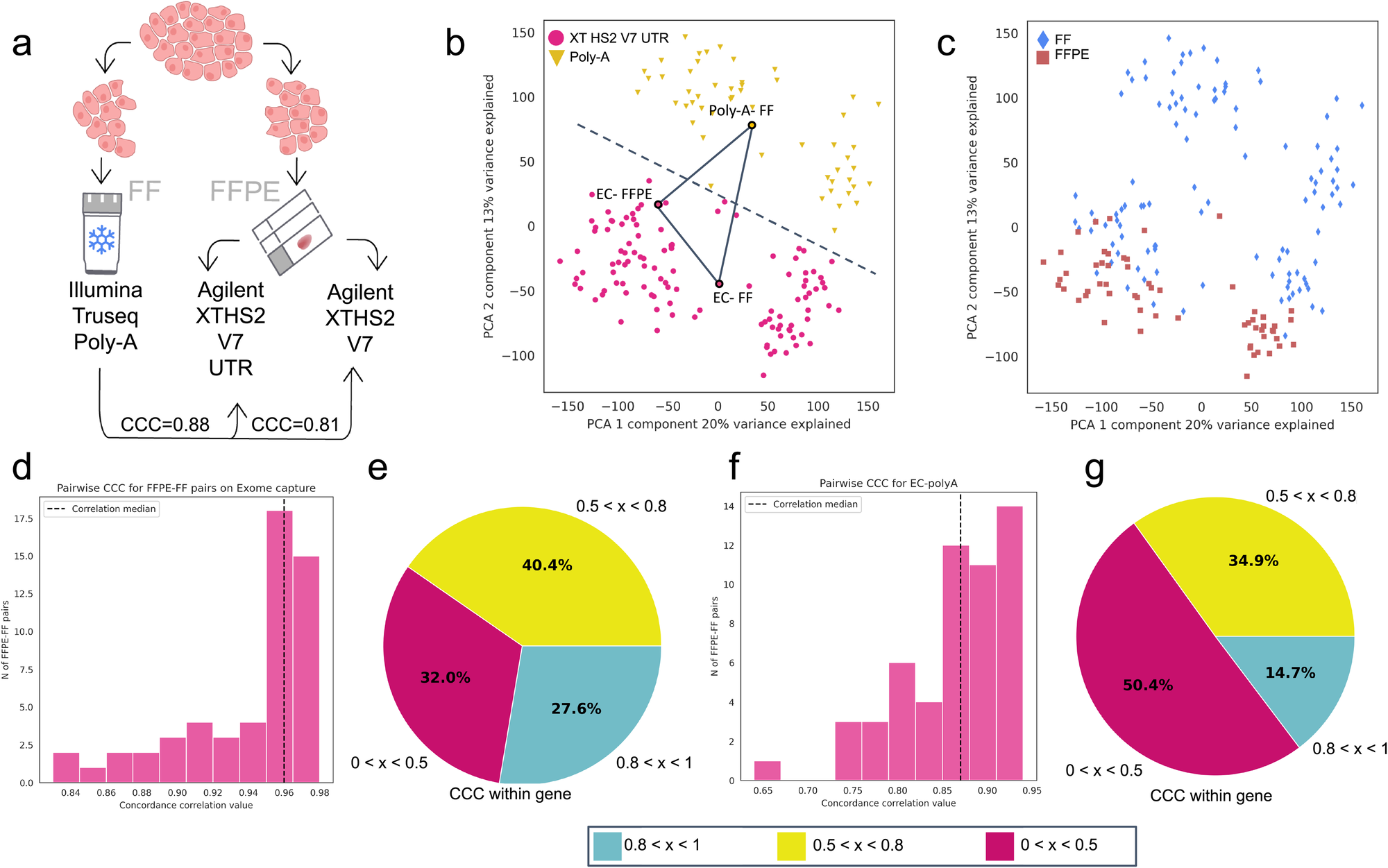
a Schematic of EC-protocol optimization. Median CCC for V7 versus V7 UTR. b Principal Component Analysis (PCA) showing batch effect between the modified EC (V7 UTR) and poly-A RNA-seq protocols. c PCA showing the absence of batch effect between samples stored as either FF or FFPE and sequenced with the modified EC protocol. d Distribution of within-sample CCC between FF and FFPE samples processed using the modified EC protocol. e Pie chart showing percentage of genes with low (<0.5), medium (0.5-0.8), and high (>0.8) within-gene CCC between samples stored as FF or FFPE and processed using the modified EC protocol. f Distribution of within-sample CCC for pairwise comparison between FF (poly-A) and FFPE (EC) samples. g Pie chart showing percentage of genes with low (<0.5), medium (0.5–0.8), and high (>0.8) within-gene CCC between modified EC and poly-A protocols. CCC concordance correlation coefficient, EC exome capture.
Applying Procrustes to samples processed using both EC and poly-A RNA-seq protocols revealed promising results. The algorithm significantly improved the correlation of gene expression between the two protocols, with 61% of genes showing strong concordance across both methods. Notably, Procrustes demonstrated superior performance in correlating cancer-specific and cancer microenvironment-related genes, crucial for accurate oncological analysis.
Furthermore, benchmarking analyses illustrated Procrustes’ superiority over other batch correction methods, highlighting its effectiveness in enhancing the accuracy and reliability of RNA-seq data. Importantly, the algorithm’s capability to project RNA-seq data from a single sample onto a larger cohort opens new avenues for comprehensive gene expression analysis in cancer research.
The implications of this study are profound. By ensuring consistency and comparability of gene expression measurements across different RNA-seq protocols, Procrustes facilitates more accurate and reliable analysis of tumor samples. This advancement paves the way for improved gene expression-based treatment decisions in personalized oncology, ultimately enhancing patient outcomes and advancing cancer research.
Availability – All code is deposited online (https://github.com/BostonGene /Procrustes)
Kotlov N, Shaposhnikov K, Tazearslan C, Chasse M, Baisangurov A, Podsvirova S, Fernandez D, Abdou M, Kaneunyenye L, Morgan K, Cheremushkin I, Zemskiy P, Chelushkin M, Sorokina M, Belova E, Khorkova S, Lozinsky Y, Nuzhdina K, Vasileva E, Kravchenko D, Suryamohan K, Nomie K, Curran J, Fowler N, Bagaev A. (2024) Procrustes is a machine-learning approach that removes cross-platform batch effects from clinical RNA sequencing data. Commun Biol 7(1):392. [article]


