

Benchmarking single-cell RNA-seq (scRNA-seq) and single-cell Assay for Transposase-Accessible Chromatin using sequencing (scATAC-seq) computational tools demands simulators to generate realistic sequencing reads. However, none of the few read simulators aim to mimic real data. To fill this gap, UCLA researchers introduce scReadSim, a single-cell RNA-seq and ATAC-seq read simulator that allows user-specified ground truths and generates synthetic sequencing reads (in a FASTQ or BAM file) by mimicking real data. At both read-sequence and read-count levels, scReadSim mimics real scRNA-seq and scATAC-seq data. Moreover, scReadSim provides ground truths, including unique molecular identifier (UMI) counts for scRNA-seq and open chromatin regions for scATAC-seq. In particular, scReadSim allows users to design cell-type-specific ground-truth open chromatin regions for scATAC-seq data generation. In benchmark applications of scReadSim, these researchers show that UMI-tools achieves the top accuracy in scRNA-seq UMI deduplication, and HMMRATAC and MACS3 achieve the top performance in scATAC-seq peak calling.
Workflows of scReadSim’s scRNA-seq and scATAC-seq read generation
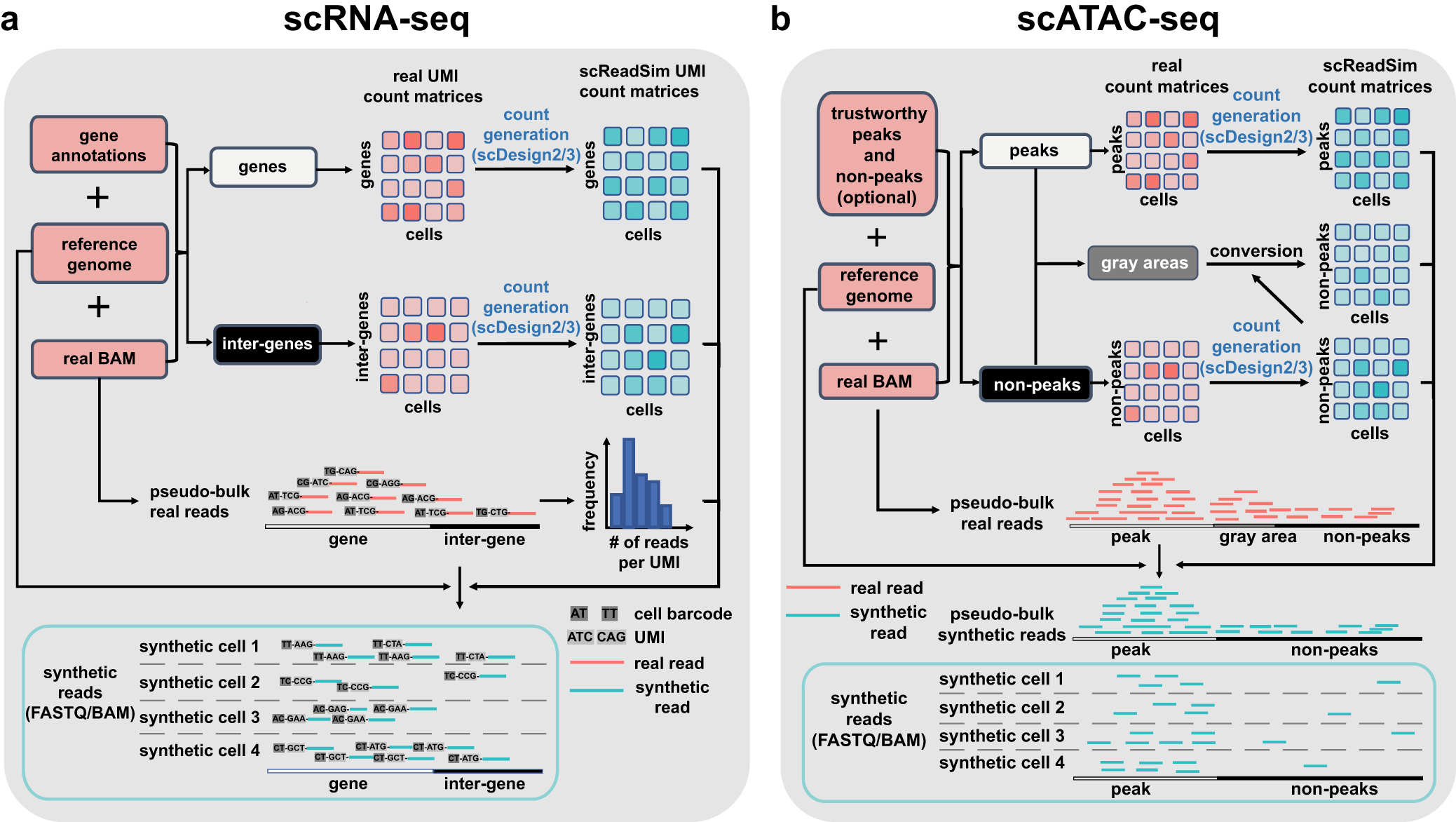
a For scRNA-seq read simulation, the required input includes a real scRNA-seq dataset’s BAM file, the corresponding reference genome, and a gene annotation GTF file. Based on the input, scReadSim segregates the reference genome into genes and inter-genes (i.e., intergenic regions). Based on the genes and inter-genes, scReadSim summarizes scRNA-seq reads in the input BAM file into a gene-by-cell UMI count matrix and an inter-gene-by-cell UMI count matrix. Then scReadSim trains the count simulator scDesign2 (if the cells belong to distinct clusters; otherwise, scDesign3 can be used if the cells follow continuous trajectories) on the two UMI count matrices to generate the corresponding synthetic UMI count matrices (“ground truths” for benchmarking UMI deduplication tools). Last, scReadSim generates synthetic scRNA-seq reads based on the synthetic UMI count matrices, the input BAM file, and the reference genome. The synthetic reads are outputted in a FASTQ or BAM file. b For scATAC-seq read simulation, the input includes a real scATAC-seq dataset’s BAM file, the corresponding reference genome, and optionally, users’ trustworthy peaks and non-peaks in the input BAM file; if users do not input trustworthy peaks and non-peaks, scReadSim provides two options; see the subsection “scReadSim for scATAC-seq” for detail. Based on the trustworthy peaks and non-peaks, scReadSim defines the complementary genomic regions as gray areas and summarizes scATAC-seq reads in the input BAM file into a peak-by-cell count matrix and a non-peak-by-cell count matrix. Next, scReadSim trains the count simulator scDesign2 (or scDesign3) on the two count matrices to generate the corresponding synthetic count matrices for the peaks and non-peaks. Further, scReadSim converts the gray areas into non-peaks (so that the peaks can be regarded as “ground-truth peaks”) and constructs a synthetic count matrix based on the gray areas’ lengths and the already-generated synthetic non-peak-by-cell count matrix. Last, scReadSim generates synthetic reads based on the three synthetic count matrices, the input BAM file, and the reference genome. The synthetic reads are outputted in a FASTQ or BAM file.
Availability – The scReadSim Python package is available at https://github.com/JSB-UCLA/scReadSim.
Yan G, Song D, Li JJ. (2023) scReadSim: a single-cell RNA-seq and ATAC-seq read simulator. Nat Commun 14(1):7482. [article]


