

Single-cell RNA sequencing has been instrumental in uncovering cellular spatiotemporal context. This task is challenging as cells simultaneously encode multiple, potentially cross-interfering, biological signals. Researchers from the Hebrew University of Jerusalem have developed scPrisma, a spectral computational method that uses topological priors to decouple, enhance and filter different classes of biological processes in single-cell data, such as periodic and linear signals. The researchers apply scPrisma to the analysis of the cell cycle in HeLa cells, circadian rhythm and spatial zonation in liver lobules, diurnal cycle in Chlamydomonas and circadian rhythm in the suprachiasmatic nucleus in the brain. scPrisma can be used to distinguish mixed cellular populations by specific characteristics such as cell type and uncover regulatory networks and cell-cell interactions specific to predefined biological signals, such as the circadian rhythm. They show scPrisma’s flexibility in incorporating prior knowledge, inference of topologically informative genes and generalization to additional diverse templates and systems. scPrisma can be used as a stand-alone workflow for signal analysis and as a prior step for downstream single-cell analysis.
General workflow of scPrisma
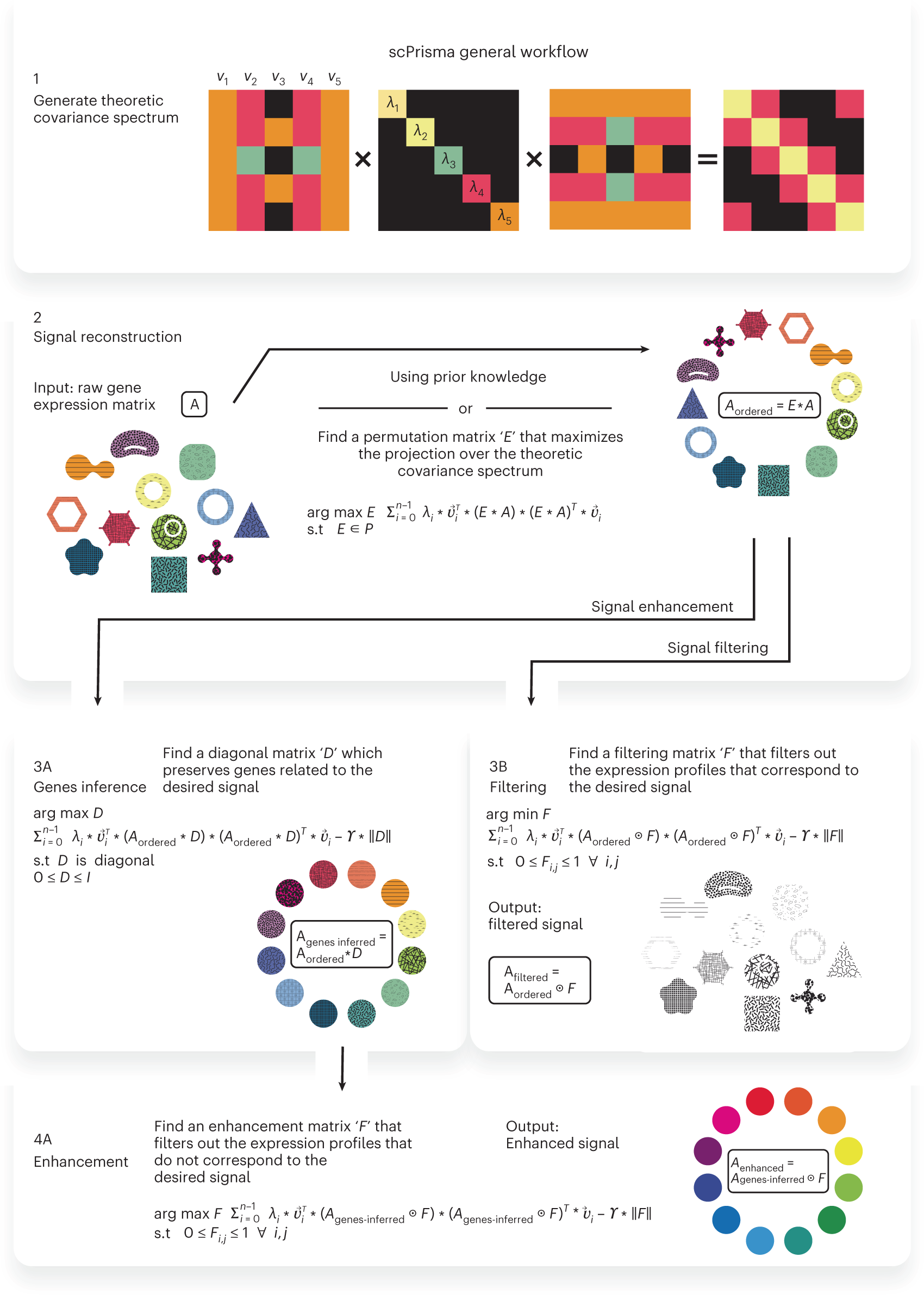
Given single-cell data and a theoretical covariance matrix, for example, based on a topological model, we calculate its theoretical spectrum (1), order the cells along the topology (2) and either enhance the signal corresponding to that topology (3A, 4A) or filter it out from the data (3B).
Availability – The code for scPrisma is publicly available at https://github.com/nitzanlab/scPrisma/
Karin J, Bornfeld Y, Nitzan M. (2023) scPrisma infers, filters and enhances topological signals in single-cell data using spectral template matching. Nat Biotechnol [Epub ahead of print]. [article]


