

Single-cell RNA-sequencing (scRNA-seq) data can serve as a good indicator of cell-to-cell heterogeneity and can aid in the study of cell growth by identifying cell types. Recently, advances in Variational Autoencoder (VAE) have demonstrated their ability to learn robust feature representations for scRNA-seq. However, it has been observed that VAEs tend to ignore the latent variables when combined with a decoding distribution that is too flexible.
Researchers at Yulin University have developed ScInfoVAE, a dimensional reduction method based on the mutual information variational autoencoder (InfoVAE), which can more effectively identify various cell types in scRNA-seq data of complex tissues. A joint InfoVAE deep model and zero-inflated negative binomial distributed model design based on ScInfoVAE reconstructs the objective function to noise scRNA-seq data and learn an efficient low-dimensional representation of it. The researchers use ScInfoVAE to analyze the clustering performance of 15 real scRNA-seq datasets and demonstrate that their method provides high clustering performance. In addition, they use simulated data to investigate the interpretability of feature extraction, and visualization results show that the low-dimensional representation learned by ScInfoVAE retains local and global neighborhood structure data well. In addition, this model can significantly improve the quality of the variational posterior.
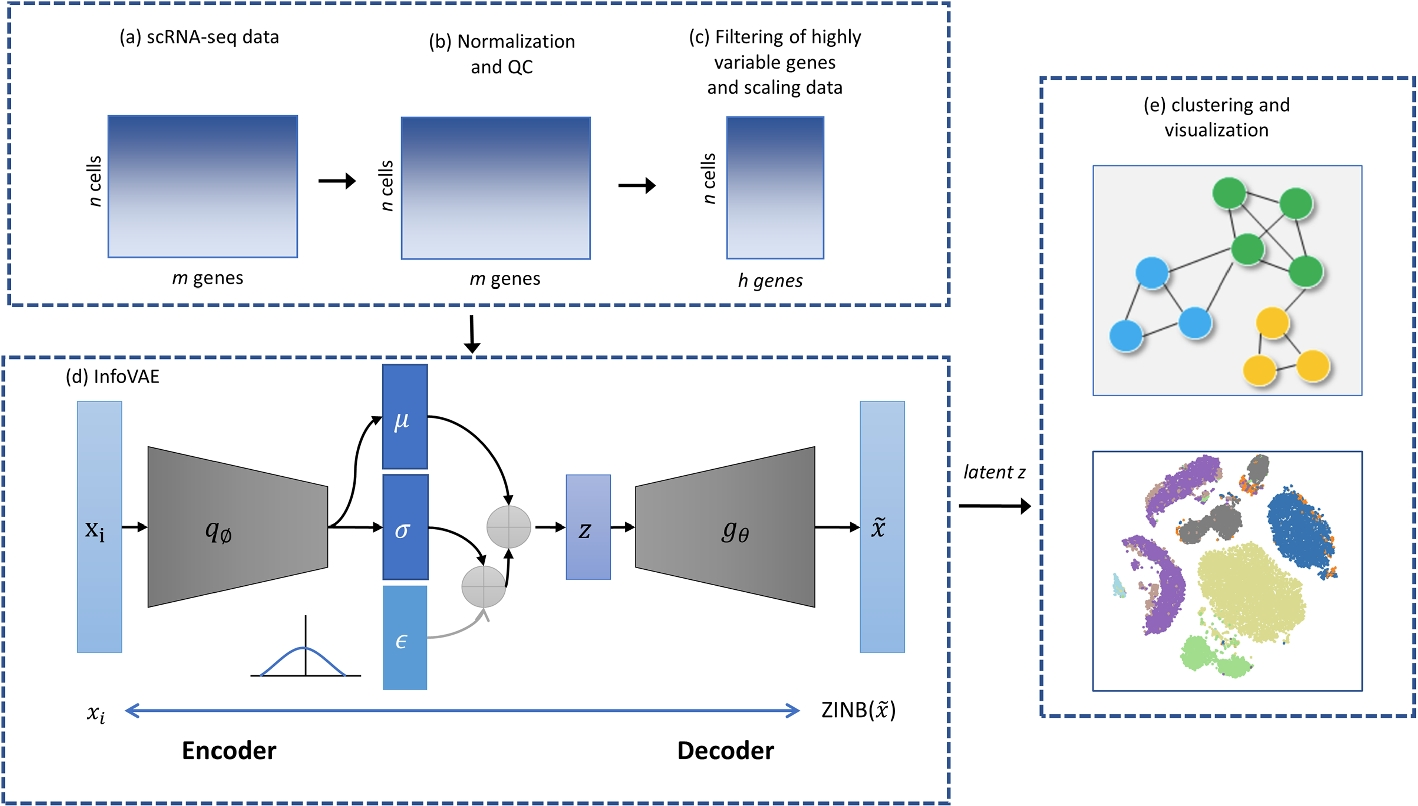
Workflow of clustering based on InfoVAE. The network is trained by both clustering loss and reconstruction loss
Pan W, Long F, Pan J. (2023) ScInfoVAE: interpretable dimensional reduction of single cell transcription data with variational autoencoders and extended mutual information regularization. BioData Min 16(1):17. [article]


