

The process of alternative splicing adds a layer of complexity to the way genetic information is expressed. This mechanism allows a single gene to produce multiple protein isoforms, vastly expanding the diversity of the transcriptome and proteome. While alternative splicing is vital for normal cellular function, its dysregulation has been implicated in numerous human diseases.
Enter RNA sequencing (RNA-seq), a powerful tool that has become the gold standard for dissecting the intricacies of alternative splicing. For nearly a decade, the Replicate Multivariate Analysis of Transcript Splicing (rMATS) software has been at the forefront of alternative splicing analysis, providing researchers with invaluable insights into transcriptomic diversity.
Now, the latest incarnation of this groundbreaking tool—rMATS-turbo—ushers in a new era of alternative splicing analysis. Developed by a dedicated team of researchers at the Children’s Hospital of Philadelphia, rMATS-turbo offers a fast and scalable approach to dissecting alternative splicing events from RNA-seq data. With its revamped computational workflow, rMATS-turbo boasts substantial improvements in speed and data storage efficiency, making it ideal for analyzing massive datasets with tens of thousands of samples.
An overview of the rMATS-turbo workflow to discover and quantify alternative splicing events in large-scale RNA-seq datasets
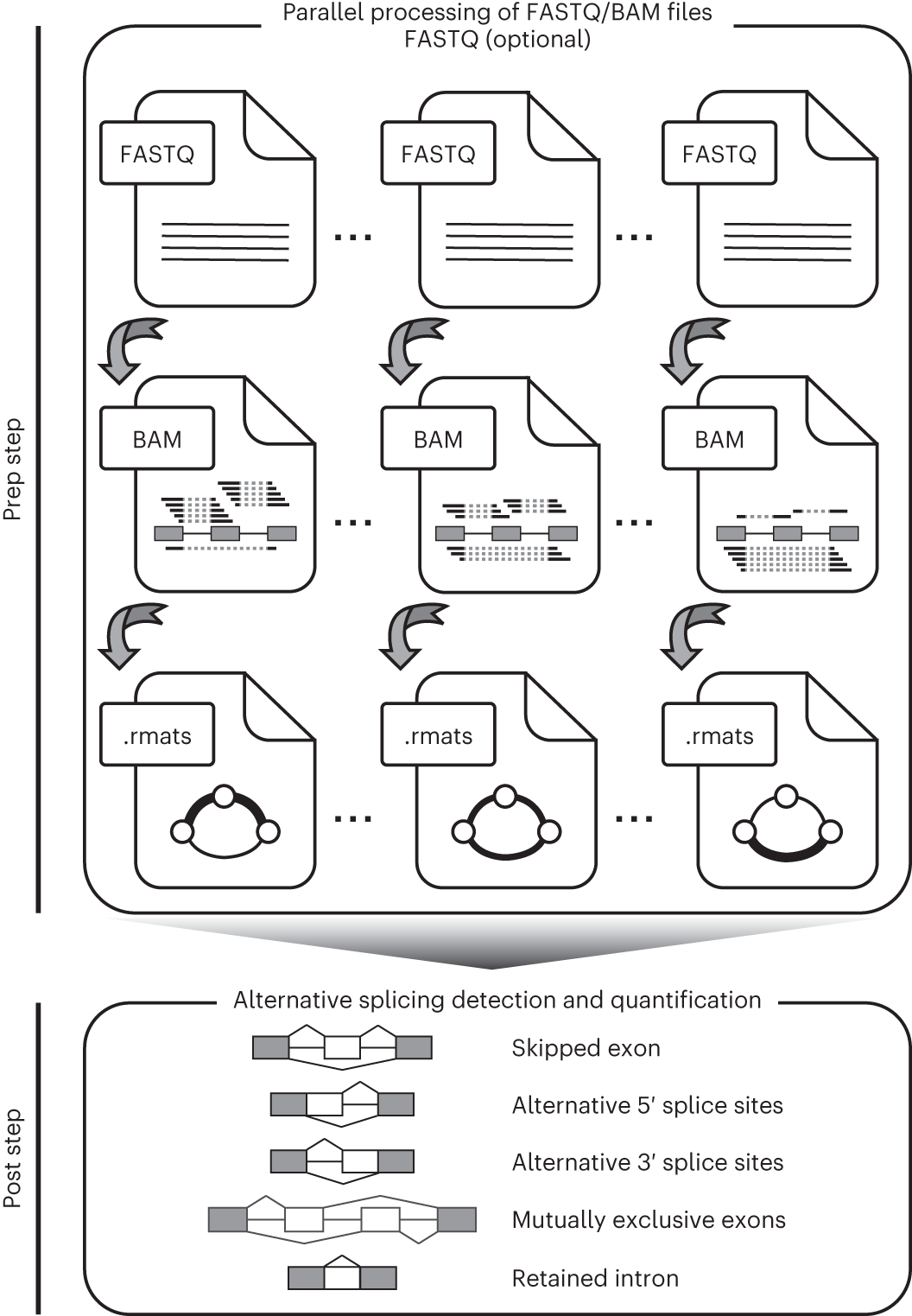
rMATS-turbo uses an efficient weighted splicing graph data structure and its associated data format (.rmats) to store splicing information extracted from raw RNA-seq data. rMATS-turbo has two main steps, ‘prep’ and ‘post’. In the prep step, input files (.FASTQ or .BAM) are processed and transformed into splicing graphs. Note that users can either start from FASTQ files or from pre-aligned BAM files. An .rmats file is saved to store information of weighted splicing graphs for each RNA-seq sample. The prep step can be run independently or in parallel on different subsets of input files. In the post step, .rmats files are read and integrated across samples to discover and quantify alternative splicing events. Five basic types of alternative splicing patterns are discovered and analyzed: SE, A5SS, A3SS, MXE and RI. The post step also incorporates a statistical model for identifying differential alternative splicing events between two sample groups.
But what sets rMATS-turbo apart? Let’s explore its key features through two illustrative application scenarios:
- Differential splicing analysis: rMATS-turbo excels in identifying differential alternative splicing events between two sample groups, whether they involve annotated variants or novel splicing events. This capability allows researchers to uncover alterations in splicing patterns associated with various physiological and pathological conditions.
- Quantitative analysis of large-scale datasets: With its ability to handle extensive RNA-seq datasets comprising thousands of samples, rMATS-turbo enables researchers to conduct quantitative analyses of alternative splicing across diverse biological systems. By delving into the intricate landscape of splicing dynamics, researchers can uncover novel insights into cellular states and disease mechanisms.
Moreover, rMATS-turbo’s efficient parallel processing capabilities enable seamless analysis on compute clusters, ensuring rapid turnaround times even for the most demanding analyses.
In conclusion, rMATS-turbo represents a significant advancement in the field of alternative splicing analysis, offering researchers an unparalleled tool for unraveling the mysteries of transcriptomic diversity. By empowering scientists to delve deeper into the complexities of alternative splicing, rMATS-turbo paves the way for groundbreaking discoveries that could revolutionize our understanding of human health and disease.
Availability – rMATS-turbo is publicly available on GitHub (https://github.com/Xinglab/rmats-turbo) and Bioconda (https://anaconda.org/bioconda/rmats)
Wang Y, Xie Z, Kutschera E. et al. (2024) rMATS-turbo: an efficient and flexible computational tool for alternative splicing analysis of large-scale RNA-seq data. Nat Protoc [Epub ahead of print]. [abstract]


