

miRNA and other forms of small RNAs are known to regulate many biological processes. Single-cell small RNA sequencing can be used to profile small RNAs of individual cells; however, limitations of efficiency and scale prevent its widespread application. Researchers at Tsinghua University have developed parallel single-cell small RNA sequencing (PSCSR-seq), which can overcome the limitations of existing methods and enable high-throughput small RNA expression profiling of individual cells. Analysis of PSCSR-seq data indicated that diverse cell types could be identified based on patterns of miRNA expression, and showed that miRNA content in nuclei is informative (for example, cell type marker miRNAs can be detected in isolated nuclei). PSCSR-seq is very sensitive: analysis of only 732 peripheral blood mononuclear cells (PBMCs) detected 774 miRNAs, whereas bulk small RNA analysis would require input RNA from approximately 106 cells to detect as many miRNAs. The researchers identified 42 miRNAs as markers for PBMC subpopulations. Moreover, they analyzed the miRNA profiles of 9,533 cells from lung cancer biopsies, and by dissecting cell subpopulations, they identified potentially diagnostic and therapeutic miRNAs for lung cancers. This study demonstrates that PSCSR-seq is highly sensitive and reproducible, thus making it an advanced tool for miRNA analysis in cancer and life science research.
High-quality single-cell small RNA sequencing with PSCSR-seq
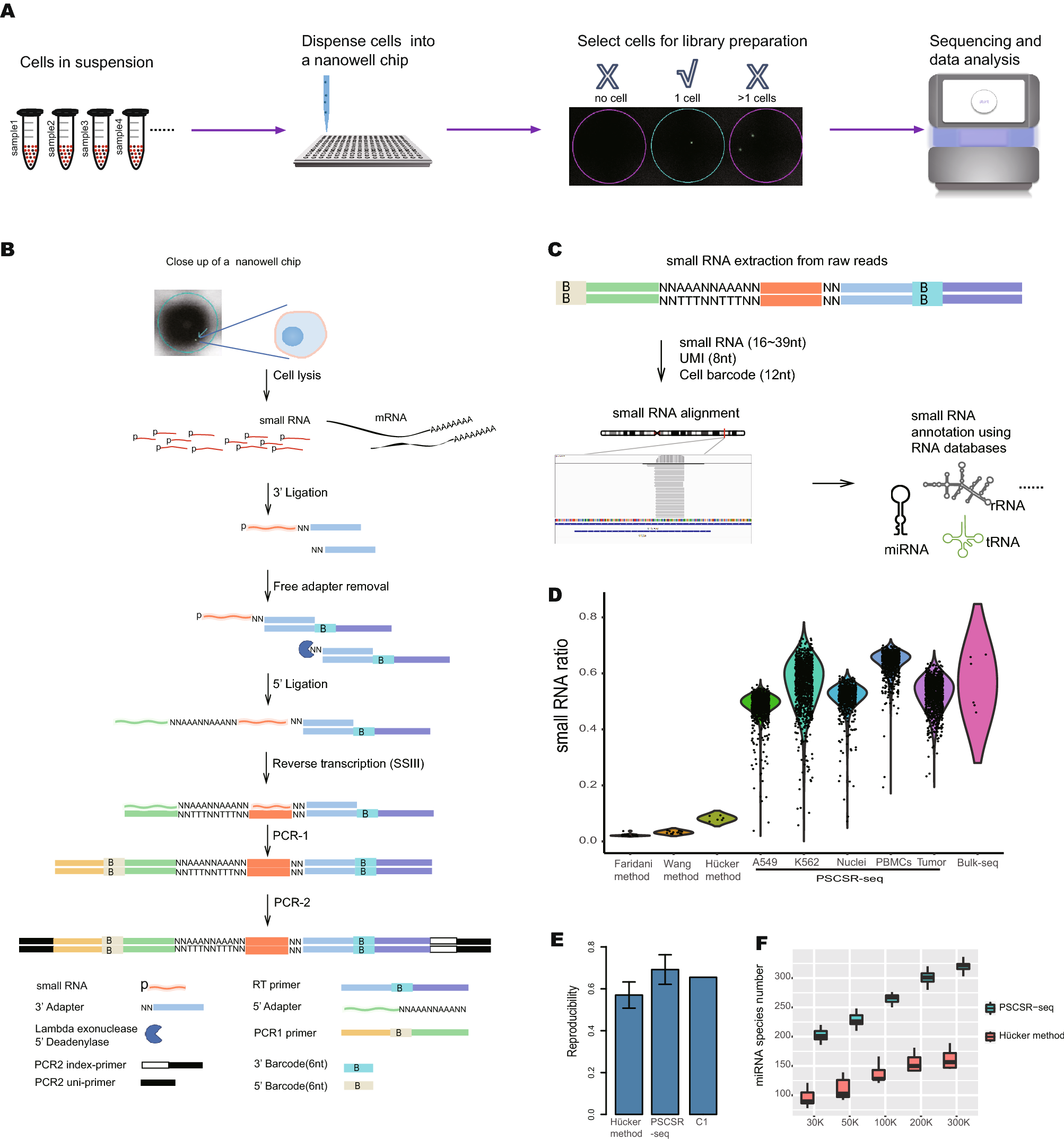
(A) Schematic of PSCSR-seq. Resuspended cells were dispersed into a nanowell chip. Individual viable cells were selected by a microscope, and small RNA libraries were prepared, followed by sequencing and data analysis. (B) Flowchart of single-cell small RNA library preparation. Cells were lysed to release small RNAs. Then, 3′ adapters were ligated to these small RNAs, and the remaining 3′ adapters were removed. Next, 5′ adapters were ligated to these small RNAs. In total, the adapters harbor 8 random nucleotides as a unique molecular index or UMI, which were used for a small RNA counting procedure during data analysis. The ligated small RNAs were then reverse transcribed. In addition, two rounds of PCR-based amplification were used to introduce barcodes for labeling cells (PCR-1) and for multiplexing of samples for high-throughput sequencing (PCR-2) (see the “PSCSR-seq library preparation and sequencing” section of Methods). (C) Small RNAs with associated UMIs and cell barcodes were extracted from the raw reads and then aligned to the genome. The small RNA read criteria were as follows: 1, the small RNA length was within 16–39 nt after removal of adapter sequences; and 2, the small RNA sequences needed to be matched to the genome without any mismatches. Small RNAs were further classified into miRNAs or tRNA-derived small RNAs, rRNA-derived small RNAs, as well as other RNA species. The expression level for each class of small RNA was then estimated (see the “PSCSR-seq data analysis” section of Methods). (D) Comparison of existing single-cell small RNA sequencing methods. We defined the small RNA ratio as the number of small RNA reads divided by the total number of sequenced reads. The small RNA reads ratios for existing methods were calculated based on published datasets (25 primed human embryonic stem cells were used with the Faridani method, 16 K562 cells were used with the Wang method, 6 A549 cells were used with the Hücker method, and 6 cancer samples with traditional bulk small RNA sequencing method, see the “comparison of methods” section of Methods). (E) Reproducibility comparison. The bar plot is based on the average reproducibility values of A549 cells (Hücker method, n = 6), A549 cells randomly chosen from PSCSR-seq results (n = 10), and the reported value from the Fluidigm C1 single-cell mRNA sequencing method. Error bars represent the standard deviation (see the “comparison of methods” section of Methods). (F) Boxplot of miRNA numbers per cell detected at different sequencing depths (random downsampling from sequences of A549 cells using the Hücker method and PSCSR-seq, n = 6 and 10 respectively
Li J, Zhang Z, Zhuang Y et al. (2023) Small RNA transcriptome analysis using parallel single-cell small RNA sequencing. Sci Rep [Epub ahead of print]. [article]


