

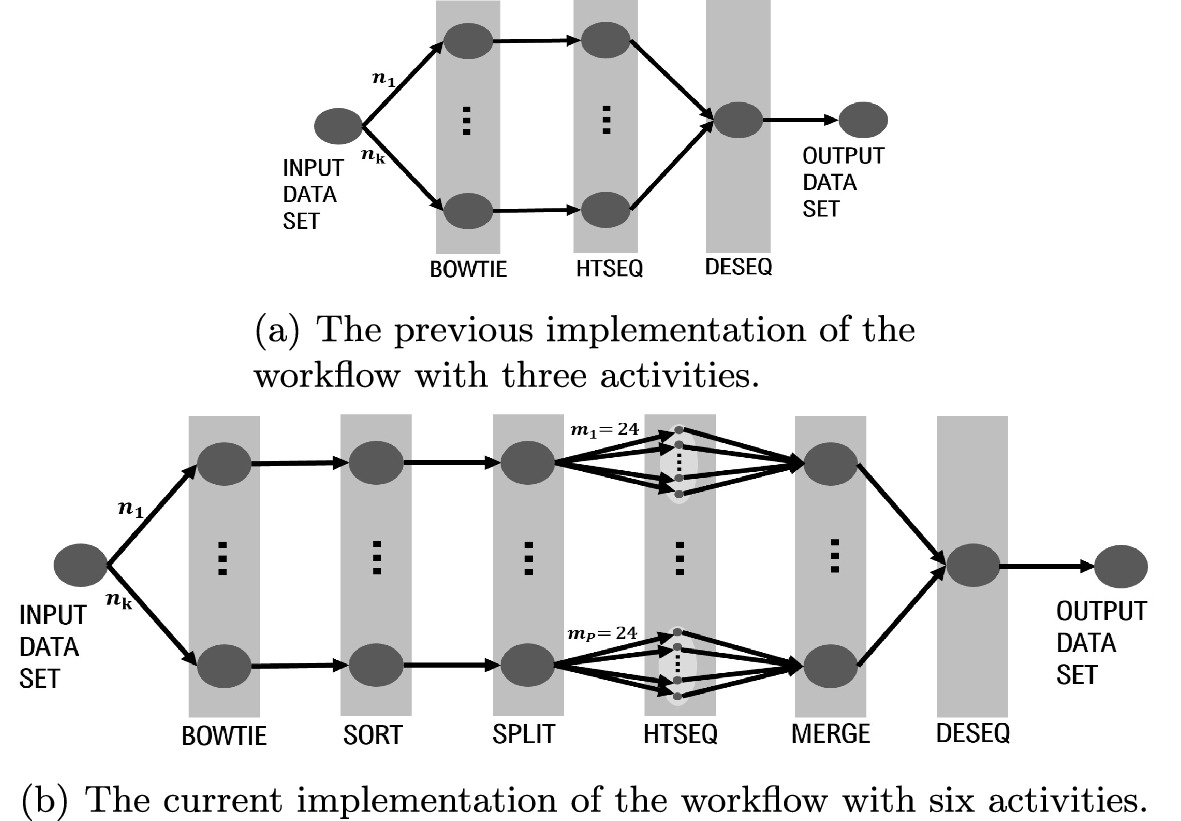
RNA sequencing has become an increasingly affordable way to profile gene expression analyses. Researchers from the LNCC, Brazil have developed a scientific workflow implementing several open-source software executed by Parsl parallel scripting language in an high-performance computing environment. The researchers have applied the workflow to a single-cardiomyocyte RNA-seq data retrieved from Gene Expression Omnibus database. The workflow allows for the analysis (alignment, QC, sort and count reads, statistics generation) of raw RNA-seq data and seamless integration of differential expression results into a configurable script code.
In this work, the researchers aim to investigate an analytical comparison of executing the workflow in Solid State Disk and Lustre as a critical decision for improving the execution efficiency and resilience in current and upcoming RNA-Seq workflows. Based on the resulting profiling of CPU and I/O data collection, they demonstrate that they can correctly identify anomalies in transcriptomics workflow performance which is an essential resource to optimize its use of high-performance computing systems. ParslRNA-Seq showed improvements in the total execution time of up to 70% against its previous sequential implementation. Finally, the researchers discuss which workflow modeling modifications lead to improved computational performance and scalability based on provenance data information.
Availability – ParslRNA-Seq is available at https://github.com/lucruzz/rna-seq
Ocaña K et al. (2022). ParslRNA-Seq: An Efficient and Scalable RNAseq Analysis Workflow for Studies of Differentiated Gene Expression. In: Navaux, P., Barrios H., C.J., Osthoff, C., Guerrero, G. (eds) High Performance Computing. CARLA 2022. Communications in Computer and Information Science, vol 1660. [abstract]


