

Single-cell RNA sequencing (scRNAseq) technologies have been beneficial in revealing and describing cellular heterogeneity within mammalian tissues, including solid tumors. However, many of these techniques apply poly(A) selection of RNA, and thus have primarily focused on determining the gene signatures of eukaryotic cellular components of the tumor microenvironment. Microbiome analysis has revealed the presence of microbial ecosystems, including bacteria and fungi, within human tumor tissues from major cancer types. Imaging data have revealed that intratumoral bacteria may be located within epithelial and immune cell types. However, as bacterial RNA typically lacks a poly(A) tail, standard scRNAseq approaches have limited ability to capture this microbial component of the tumor microenvironment. To overcome this, Fred Hutchinson Cancer Center researchers have developed the invasion-adhesion-directed expression sequencing (INVADEseq) approach, whereby they adapt 10x Genomics 5′ scRNAseq protocol by introducing a primer that targets a conserved region of the bacterial 16S ribosomal RNA gene in addition to the standard primer for eukaryotic poly(A) RNA selection. This ‘add-on’ approach enables the generation of eukaryotic and bacterial DNA libraries at eukaryotic single-cell level resolution, utilizing the 10x barcode to identify single cells with intracellular bacteria. The INVADEseq method takes 30 h to complete, including tissue processing, sequencing and computational analysis. As an output, INVADEseq has shown to be a reliable tool in human cancer cell lines and patient tumor specimens by detecting the proportion of human cells that harbor bacteria and the identities of human cells and intracellular bacteria, along with identifying host transcriptional programs that are modulated on the basis of associated bacteria.
Tumor processing for single-cell RNAseq acquisition and computational pipeline
for host and bacteria cell annotations, host-associated transcriptome
and GSEA pathway enrichment analysis
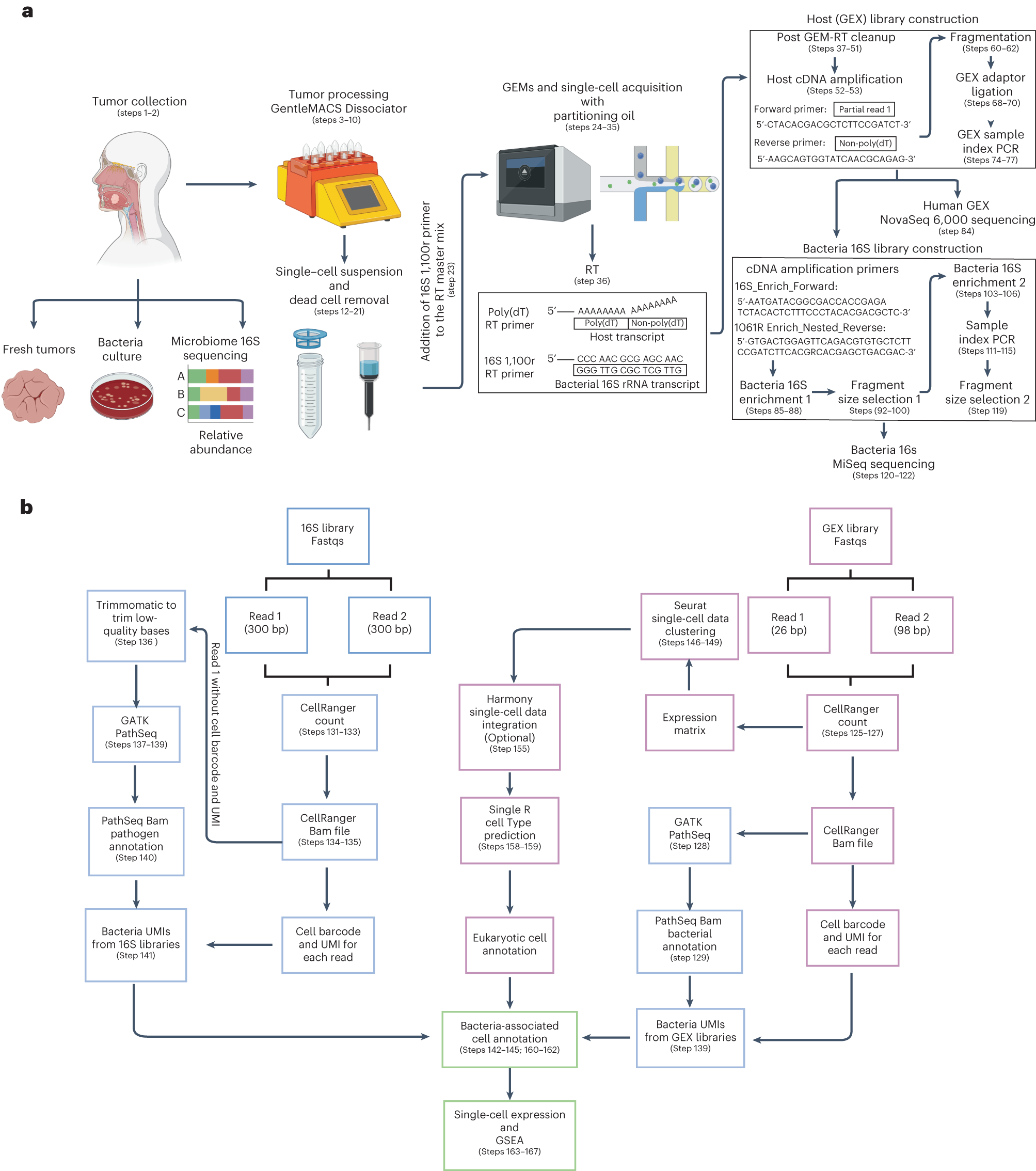
a, Tumor samples were isolated from patients with gastrointestinal tract cancers. Bacteria culture in blood agars and microbiome 16S rRNA sequencing analysis were performed to screen tumor samples that were positive for bacteria. To obtain single-cell suspensions, tumor samples were processed using the gentleMACS-quality Octo Dissociator equipped with electrical heaters. The cell suspension was passed through a 70-µm cell strainer and dead cells were removed by magnetic sorting using LS columns. Single-cell suspensions were loaded onto a Chromium Chip K and processed with the 10x Chromium controller to capture single cells within a gel bead emulsion (GEM) containing a master mix with two primers, one that targets the polyadenylated host mRNA and second that targets the bacterial 16S rRNA gene. Following RT, the hosts (GEX) cDNA libraries were prepared and sequenced using the NovaSeq 600 platform. An aliquot from the GEX cDNA libraries were acquired to enriched for bacteria transcripts by amplifying the bacterial 16S rRNA gene. Using the BluePippin system fragment sizes between 955 and 1,215 bp were selected generating the bacteria 16S libraries that were sequenced using the MiSeq platform. b, Reads from the GEX libraries were mapped with the human reference genome GRCh38 using Cellranger Count. Then, the unmapped GEX reads with an adequate cell barcode and UMI count were processed via GATK PathSeq, thus obtaining bacterial UMI matrices for each bacteria-associated single cell. Reads from the 16S bacterial enrichment libraries were processed using Cellranger Count to obtain the corresponding barcode and UMI. Then R1 reads without a barcode or UMI were trimmed to remove low-quality bases and converted to BAM files to process through GATK PathSeq obtaining the bacteria UMI matrix for valid host cells from the GEX libraries. The bacterial UMI matrices from the GEX and 16S bacterial enrichment libraries were merged, removing UMI duplicates. Single-cell expression matrices from the GEX libraries were processed by Seurat followed by SingleR package software to obtain the annotations for each eukaryotic cell cluster. Harmony software was used to integrate single-cell datasets when it was required. The merged bacteria matrix was attached to the single-cell data identifying the host single cells that harbored bacterial transcripts. Gene expression profile and GSEA pathway enrichment analyses were performed based on the presence or absence of bacteria, at various taxonomic levels, at host single-cell-level resolution.
Availability – Code for data processing and analysis of single-cell RNAseq data is available at https://github.com/FredHutch/Galeano-Nino-Bullman-Intratumoral-Microbiota_2022.
Galeano Niño JL, Wu H, LaCourse KD, Srinivasan H, Fitzgibbon M, Minot SS, Sather C, Johnston CD, Bullman S. (2023) INVADEseq to identify cell-adherent or invasive bacteria and the associated host transcriptome at single-cell-level resolution. Nat Protoc [Epub ahead of print]. [article]


