

Accurate and timely monitoring of the evolution of SARS-CoV-2 is crucial for identifying and tracking potentially more transmissible/virulent viral variants, and implement mitigation strategies to limit their spread. Researchers at the Consiglio Nazionale delle Ricerche have developed HaploCoV, a novel software framework that enables the exploration of SARS-CoV-2 genomic diversity through space and time, to identify novel emerging viral variants and prioritize variants of potential epidemiological interest in a rapid and unsupervised manner. HaploCoV can integrate with any classification/nomenclature and incorporates an effective scoring system for the prioritization of SARS-CoV-2 variants. By performing retrospective analyses of more than 11.5 M genome sequences the researchers show that HaploCoV demonstrates high levels of accuracy and reproducibility and identifies the large majority of epidemiologically relevant viral variants – as flagged by international health authorities – automatically and with rapid turn-around times. These results highlight the importance of the application of strategies based on the systematic analysis and integration of regional data for rapid identification of novel, emerging variants of SARS-CoV-2. The researchers believe that the approach outlined in this study will contribute to relevant advances to current and future genomic surveillance methods.
Workflow and potential applications of HaploCoV
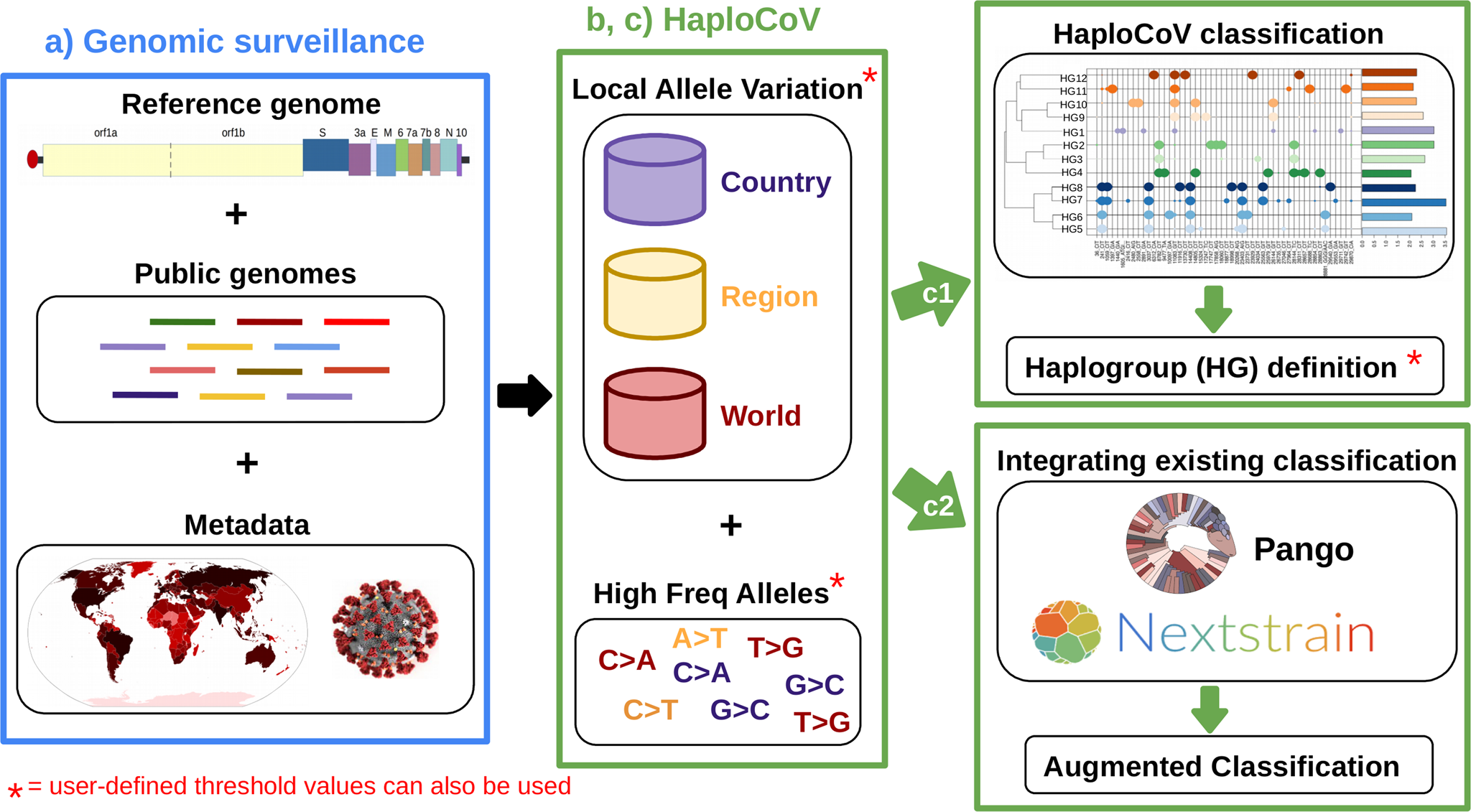
a SARS-CoV-2 genomic surveillance: genome sequences and associated metadata, obtained from publicly available repositories and/or other resources, are consolidated into a local database. b HaploCoV workflow: Firstly, genomic sequences are compared with the reference genome assembly of SARS-CoV-2 to derive a complete collection of genomic variants. Subsequently allele frequencies are computed and a collection of high frequency genomic variants is obtained. Finally, phenetic clustering of high frequency genomic variants is applied: to (c1) derive HGs of SARS-CoV-2 based on a user defined minimum phenetic distance (i.e., groups that differ by more than a user defined number of genomic variants); and/or (c2) to complement an existing classification system by applying phenetic clustering to pre-defined groups/lineages.
Availability – The software suite for analysis and classification of SARS-CoV-2 genomes described in study is available at: https://github.com/matteo14c/HaploCoV and Zenodo.
Chiara M, Horner DS, Ferrandi E, Gissi C, Pesole G. (2023) HaploCoV: unsupervised classification and rapid detection of novel emerging variants of SARS-CoV-2. Commun Biol 6(1):443. [article]


