

The cost and complexity of generating a complete reference genome means that many organisms lack an annotated reference. An alternative is to use a de novo reference transcriptome. This technology is cost-effective but is susceptible to off-target RNA contamination. Researchers at the National Center for Biotechnology Information have developed GTax, a taxonomy-structured database of genomic sequences that can be used with BLAST to detect and remove foreign contamination in RNA sequencing samples before assembly. In addition, the researchers use a de novo transcriptome assembly of Solanum lycopersicum (tomato) to demonstrate that removing foreign contamination in sequencing samples reduces the number of assembled chimeric transcripts.
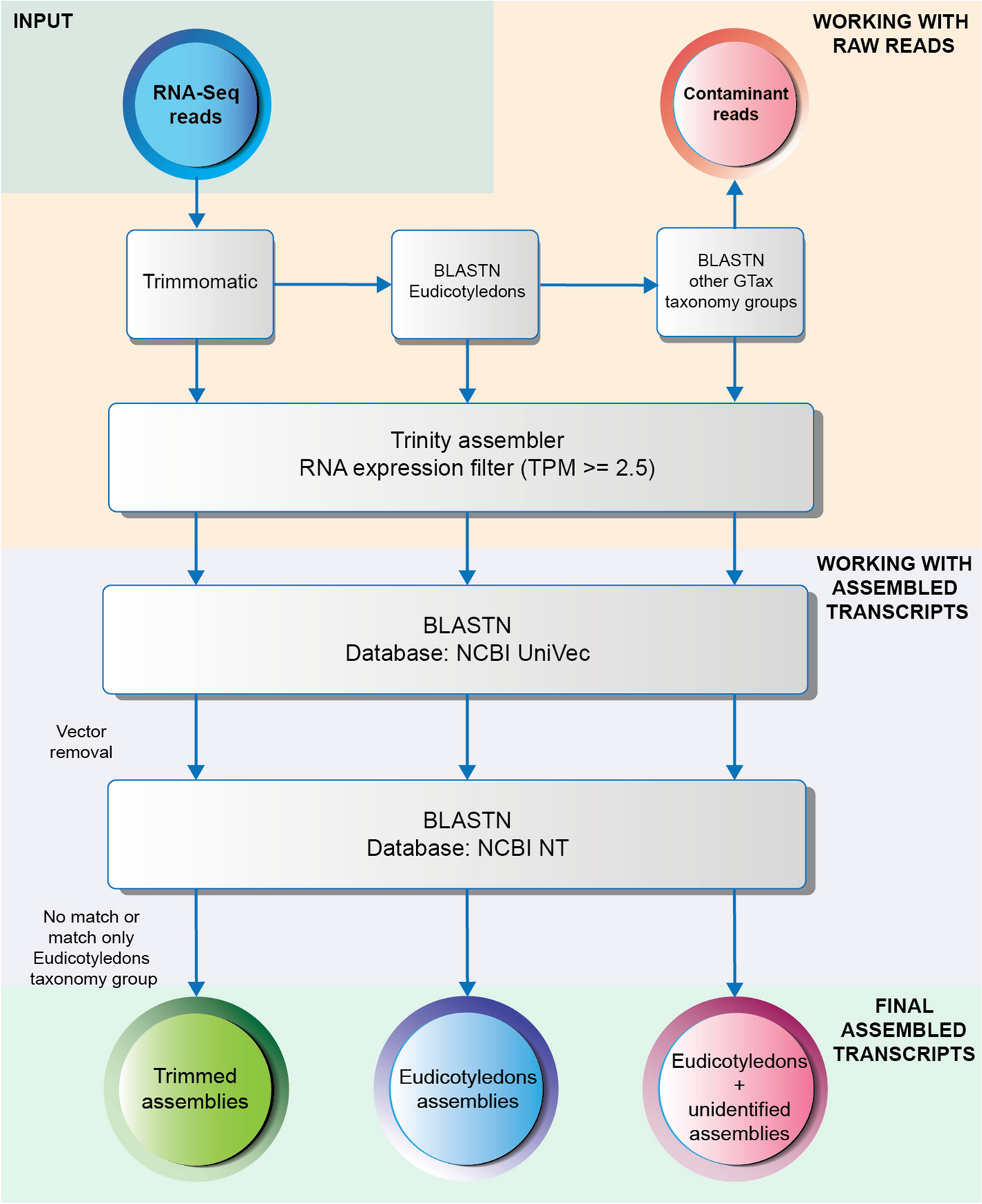
Workflow to remove vectors and contaminated transcripts after assembly completion. Different levels of decontamination of the SRA samples were used to assemble three transcriptomes: Trimmed, Eudicotyledons, and Eudicotyledons + unidentified
Availability – GTax is implemented as a Python package under Public Domain license. Source code is available at https://github.com/ncbi/gtax
Alvarez RV, Landsman D. (2024) GTax: improving de novo transcriptome assembly by removing foreign RNA contamination. Genome Biol 25(1):12. [article]


