

Spatial transcriptomics maps gene expression across tissues, posing the challenge of determining the spatial arrangement of different cell types. However, spatial transcriptomics spots contain multiple cells. Therefore, the observed signal comes from mixtures of cells of different types. Here, researchers from the Warsaw University of Technology propose an innovative probabilistic model, Celloscope, that utilizes established prior knowledge on marker genes for cell type deconvolution from spatial transcriptomics data. Celloscope outperforms other methods on simulated data, successfully indicates known brain structures and spatially distinguishes between inhibitory and excitatory neuron types based in mouse brain tissue, and dissects large heterogeneity of immune infiltrate composition in prostate gland tissue.
Celloscope overview
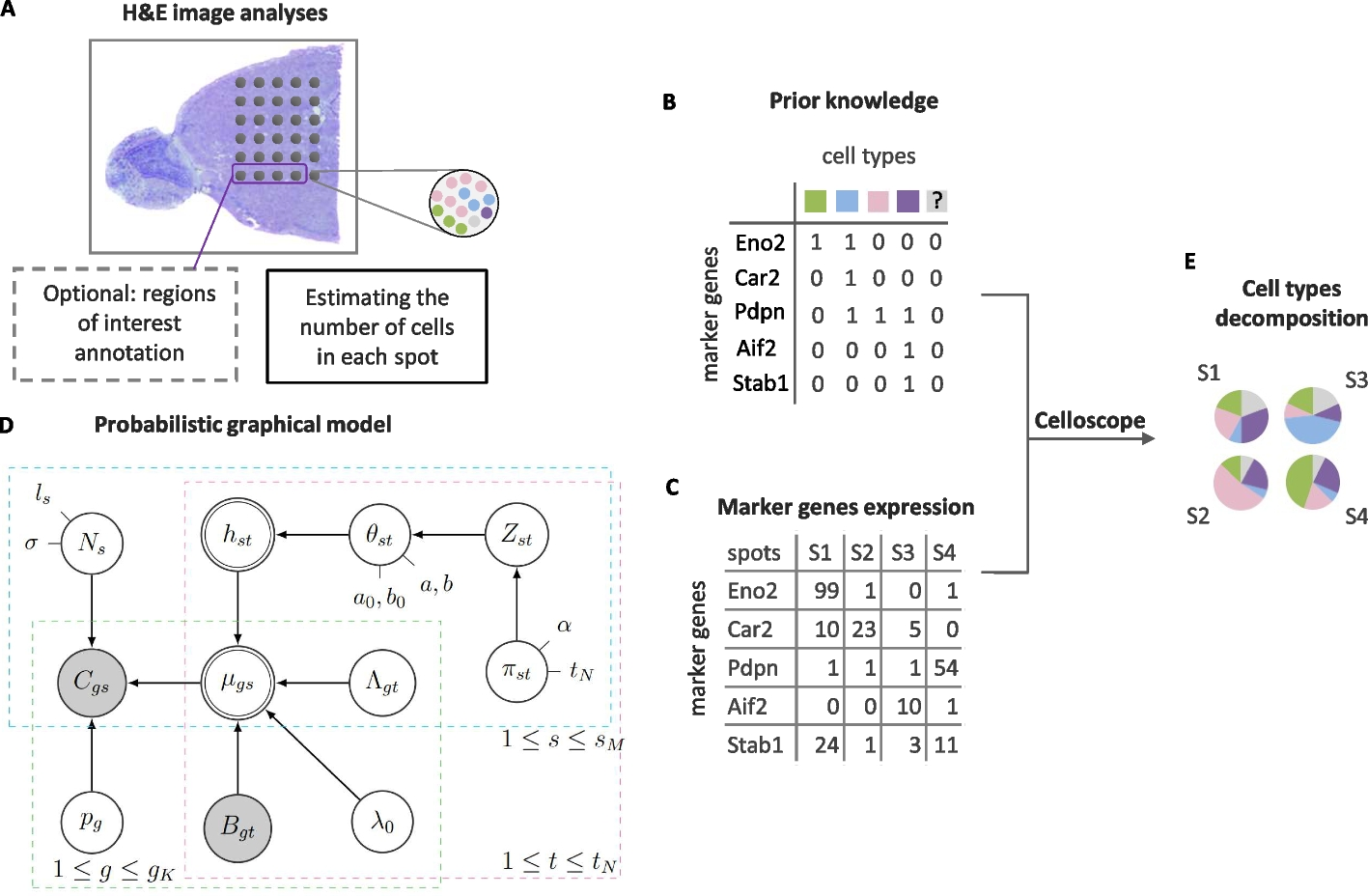
A The total number of cells for each spot is estimated based on a H&E image. Optionally, regions of interest are annotated. H&E image source: 10x Genomics. B Prior knowledge on marker genes is given as the model’s input, in a form of a binary matrix, together with ST data on marker gene expression in spots (C). D The graphical representation of Celloscope. Gray nodes correspond to the observed variables, double circled to deterministic ones, while the remaining nodes correspond to hidden variables. Arrows represent probabilistic dependencies. Model’s variables are described in Table 1 and hyperparameters in Table 2. E Cell type decomposition in each spot using Celloscope is performed via MCMC inference
Availability – https://github.com/szczurek-lab/Celloscope and https://zenodo.org/record/7817712
Geras A, Darvish Shafighi S, Domżał K, Filipiuk I, Rączkowski Ł, Szymczak P, Toosi H, Kaczmarek L, Koperski Ł, Lagergren J, Nowis D, Szczurek E. (2023) Celloscope: a probabilistic model for marker-gene-driven cell type deconvolution in spatial transcriptomics data. Genome Biol 24(1):120. [article]


