

Single-cell RNA sequencing (scRNA-seq) has revolutionized our understanding of cellular heterogeneity in health and disease. However, the lack of physical relationships among dissociated cells has limited its applications. To address this issue, researchers from McGill University and the Perelman School of Medicine, University of Pennsylvania have developed CeLEry (Cell Location recovEry), a supervised deep learning algorithm that leverages gene expression and spatial location relationships learned from spatial transcriptomics to recover the spatial origins of cells in scRNA-seq. CeLEry has an optional data augmentation procedure via a variational autoencoder, which improves the method’s robustness and allows it to overcome noise in scRNA-seq data. The researchers show that CeLEry can infer the spatial origins of cells in scRNA-seq at multiple levels, including 2D location and spatial domain of a cell, while also providing uncertainty estimates for the recovered locations. Comprehensive benchmarking evaluations on multiple datasets generated from brain and cancer tissues using Visium, MERSCOPE, MERFISH, and Xenium demonstrate that CeLEry can reliably recover the spatial location information for cells using scRNA-seq data.
Workflow of CeLEry
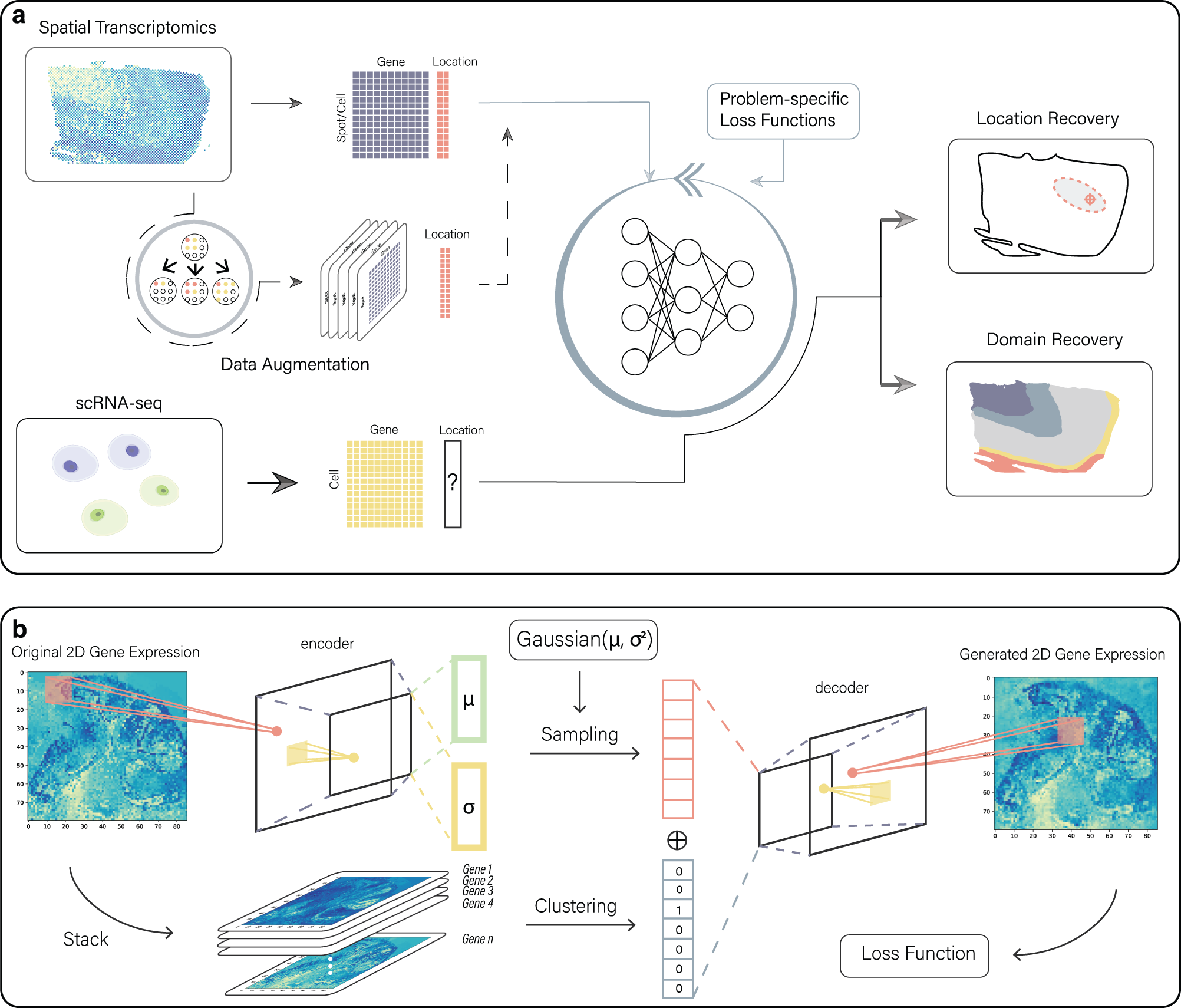
a CeLEry takes an ST dataset as input for model training and a scRNA-seq dataset as input for cell location prediction. CeLEry has an optional data augmentation step, which optionally generates replicates of the ST data via a variational autoencoder. The generated data are then included in the training data. A deep neural network is trained to learn the relationship between the spot-wise gene expression and location information by minimizing a loss function that is specified according to the specific problem. Then, the trained model is applied to the scRNA-seq data to predict the location of each cell. b The data augmentation procedure consists of an encoding stage and a decoding stage. In the encoding stage, the 2D expression pattern for each gene is summarized into the embeddings of mean and standard error vectors of a multivariate normal distribution. In the decoding stage, new embeddings are generated via the multivariate normal distribution given by the encoding stage. Meanwhile, a clustering algorithm is performed to cluster genes into groups. Finally, the generated embedding and the gene cluster embedding are concatenated, which is used as input for a convolutional neural network to decode the concatenated embedding into a 2D matrix with the same dimension as the gene expression input. The resulting 2D matrix can generate replicates of the ST data.
Zhang Q, Jiang S, Schroeder A, Hu J, Li K, Zhang B, Dai D, Lee EB, Xiao R, Li M. (2023) Leveraging spatial transcriptomics data to recover cell locations in single-cell RNA-seq with CeLEry. Nat Commun 14(1):4050. [article]


