

Integration of single-cell RNA sequencing data between different samples has been a major challenge for analyzing cell populations. However, strategies to integrate differential expression analysis of single-cell data remain underinvestigated. Researchers from the Ulsan National Institute of Science and Technology, Korea benchmark 46 workflows for differential expression analysis of single-cell data with multiple batches. The researchers show that batch effects, sequencing depth and data sparsity substantially impact their performances. Notably, they found that the use of batch-corrected data rarely improves the analysis for sparse data, whereas batch covariate modeling improves the analysis for substantial batch effects. They show that for low depth data, single-cell techniques based on zero-inflation model deteriorate the performance, whereas the analysis of uncorrected data using limmatrend, Wilcoxon test and fixed effects model performs well. The researchers suggest several high-performance methods under different conditions based on various simulation and real data analyses. Additionally, they demonstrate that differential expression analysis for a specific cell type outperforms that of large-scale bulk sample data in prioritizing disease-related genes.
An overview of our benchmark study for differential expression (DE)
analysis of scRNA-seq data with multiple batches
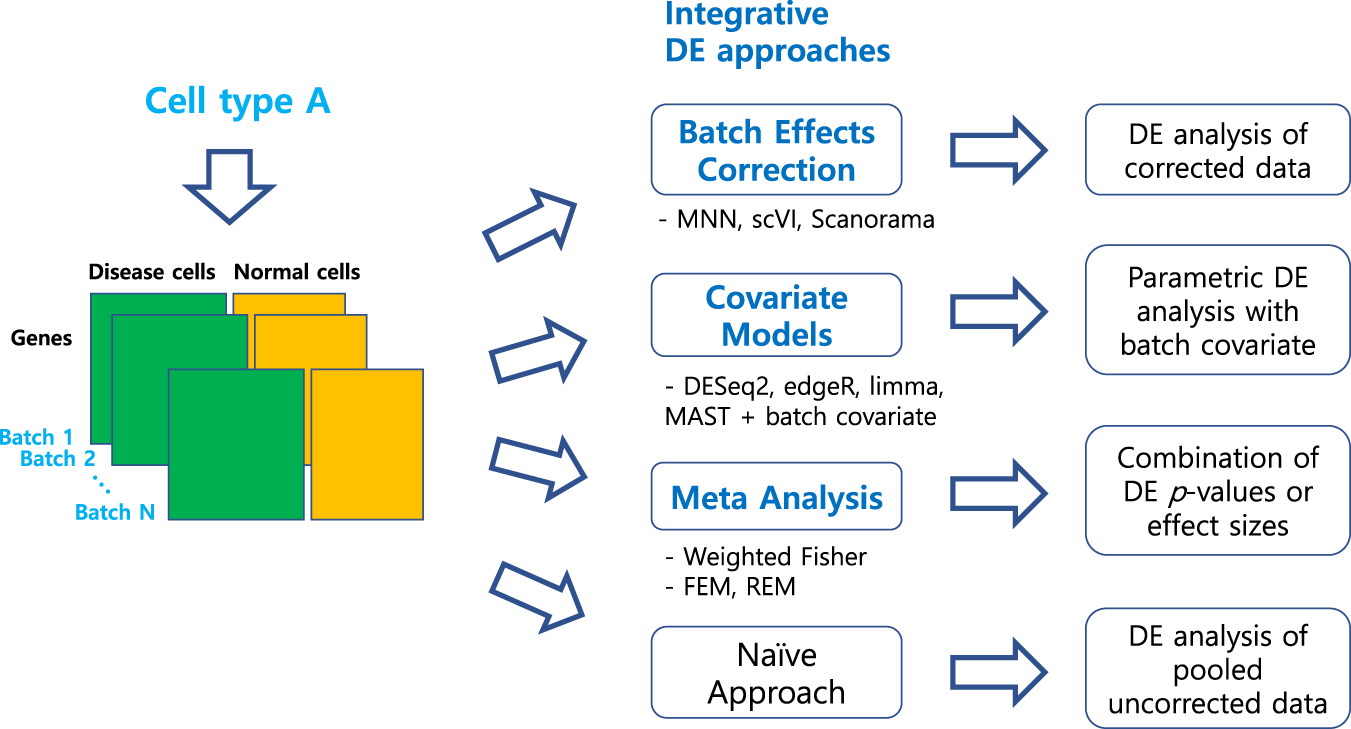
In total, 46 workflows from three integrative strategies and the naïve approach were tested.
Nguyen HCT, Baik B, Yoon S, Park T, Nam D. (2023) Benchmarking integration of single-cell differential expression. Nat Commun 14(1):1570. [article]


