

In the ever-evolving landscape of cancer research, scientists are constantly seeking innovative approaches to classify and understand different types of cancer. One promising avenue is the use of RNA sequencing (RNA-Seq) combined with machine learning techniques, which offers a modern and powerful tool for molecular classification.
However, a significant challenge in this field is the integration of molecular datasets from various sources, such as different labs or studies. These datasets often vary in quality, collection methods, and contain unwanted noise, which can hinder the accuracy of predictive models.
To address this challenge, researchers have explored the impact of data preprocessing steps on the performance of cancer classification models. These preprocessing steps include normalization, batch effect correction, and data scaling, all aimed at reducing systematic variations and harmonizing the datasets before constructing predictive models.
In a recent study, researchers at the University of Nevada Las Vegas aimed to improve the cross-study predictions of tissue of origin for common cancers using large-scale RNA-Seq datasets derived from thousands of patients across multiple tumor types. The results of their investigation highlighted the significant impact of data preprocessing operations on the performance of classifier models.
Using The Cancer Genome Atlas (TCGA) as a training set, the researchers found that batch effect correction led to improved performance in resolving tissue of origin when compared against an independent test dataset from the Genotype-Tissue Expression (GTEx) project. However, the application of data preprocessing operations resulted in worsened classification performance when the independent test dataset was aggregated from separate studies in the International Cancer Genome Consortium (ICGC) and Gene Expression Omnibus (GEO).
Flow chart of data preprocessing, machine learning, and evaluation approaches
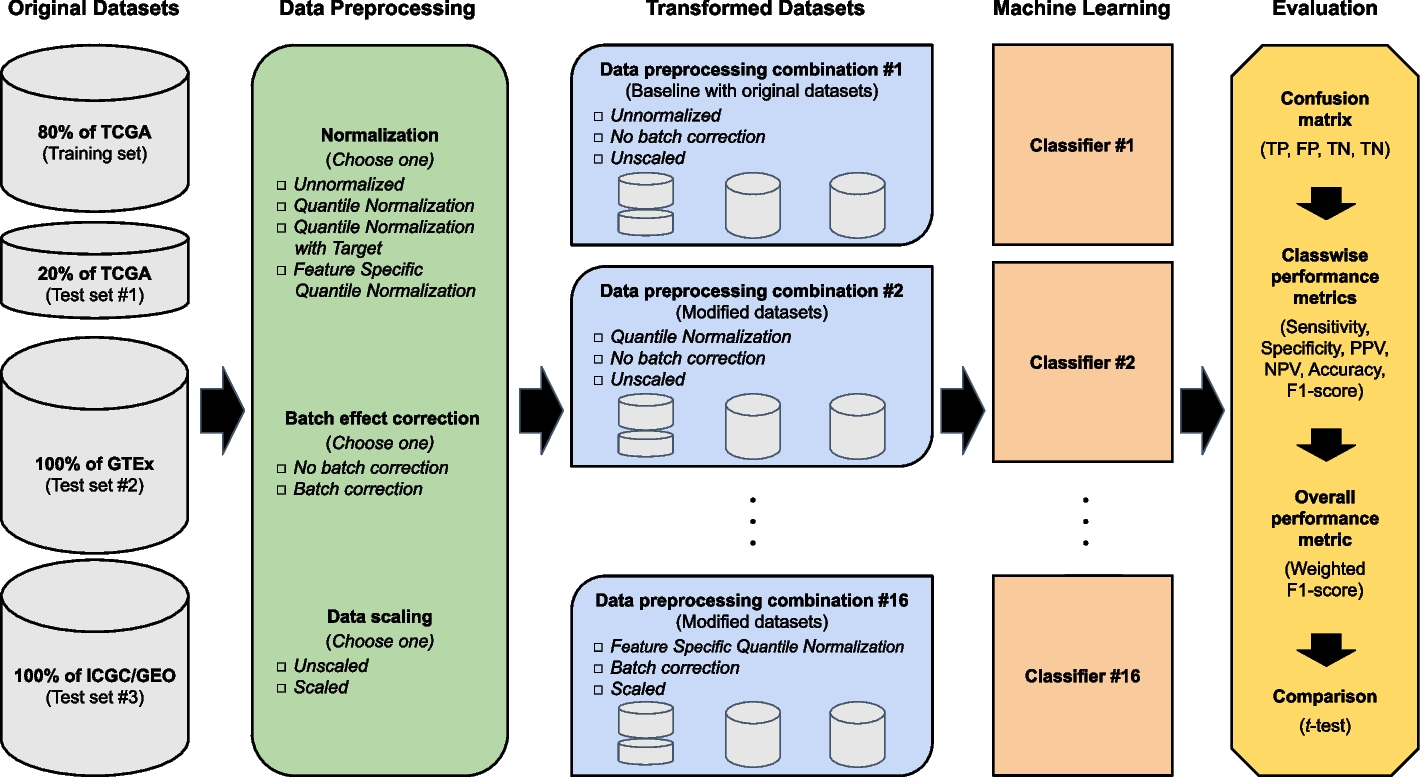
Large-scale RNA-Seq datasets were freely available to be obtained from TCGA, GTEx, ICGC, and GEO and assigned to training and test sets. The original datasets are part of data preprocessing combination #1 and serve as the ‘baseline’ for comparison. The transformed datasets of data preprocessing combinations #2 through #16 are based on various combinations of normalization (Unnormalized, Quantile Normalization, Quantile Normalization with Target, and Feature Specific Quantile Normalization), batch effect correction (No batch effect correction or Batch effect correction), and data scaling (Unscaled or Scaled) procedures applied to the original datasets. Each of the data preprocessing combinations is used to build an associated machine learning classifier
These findings underscore the complexity of integrating and analyzing large-scale RNA-Seq datasets for cancer classification. While data preprocessing techniques can enhance performance in certain scenarios, they may not always be appropriate, particularly when datasets are aggregated from diverse sources.
Overall, this study sheds light on the importance of carefully considering data preprocessing steps in RNA sequencing analysis for cancer classification. By understanding how these preprocessing techniques impact predictive models, researchers can improve the accuracy and reliability of cancer classification methods, ultimately advancing our ability to diagnose and treat cancer effectively.
Van R, Alvarez D, Mize T et al. (2024) A comparison of RNA-Seq data preprocessing pipelines for transcriptomic predictions across independent studies. BMC Bioinformatics [Epub ahead of print]. [article]


