

In the intricate landscape of genomics, uncovering structural variations (SVs) within the genome is akin to searching for hidden treasure. These SVs, which include deletions, duplications, and rearrangements of DNA segments, play a crucial role in understanding genetic diseases and evolutionary processes. However, calling SVs accurately has long been a daunting task for researchers.
Enter Sniffles2, a cutting-edge tool that promises to revolutionize SV calling by leveraging long-read sequencing technology. In a recent study, researchers at the Human Genome Sequencing Center Baylor College of Medicine unveiled the power of Sniffles2 in identifying complex genomic alterations with unprecedented accuracy and efficiency.
Overview of Sniffles2
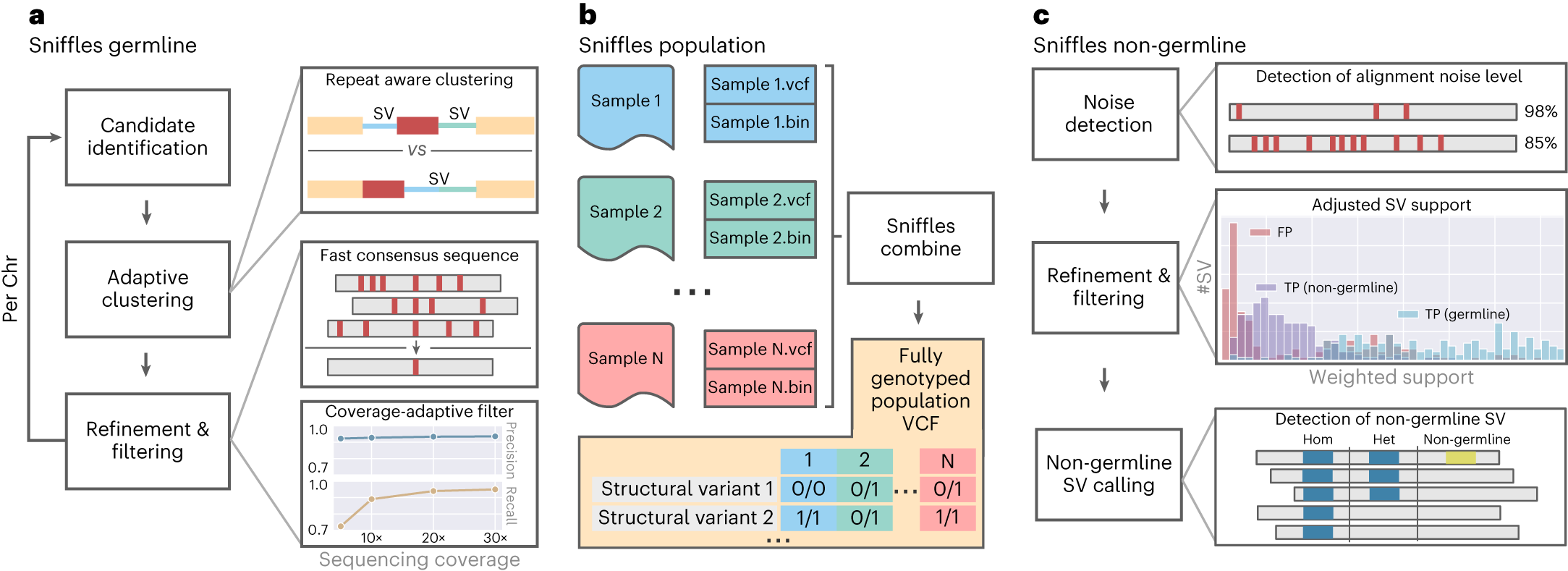
a, For Sniffles2, the researchers implemented a repeat aware clustering coupled with a fast consensus sequence and coverage-adaptive filtering to improve accuracy of the germline SV calls. b, One key limitation of current SV calling is the generation of fully genotyped population VCF. Sniffles2 implements a concept similar to a gVCF file where single-sample calling is done only once, which reduces runtime multiple-fold. c, Mosaic SV detection is enabled by improved detection and filtering of low VAF SVs (by default, 5–20%) across a bulk sample. This is enabled over additional noise detection methodology as well as refinement and filtering approaches that they developed.
One of the main challenges in SV calling lies in distinguishing true SVs from sequencing artifacts and repetitive regions in the genome. Sniffles2 addresses this challenge head-on by implementing a repeat-aware clustering algorithm, coupled with a fast consensus sequence and coverage-adaptive filtering. This innovative approach not only improves the accuracy of SV detection but also significantly speeds up the process, making it 11.8 times faster than current state-of-the-art SV callers.
But speed is not the only advantage of Sniffles2. The tool boasts a remarkable 29% increase in accuracy across various coverages (ranging from 5 to 50 times), sequencing technologies (such as Oxford Nanopore Technology and HiFi sequencing), and SV types. This means that researchers can now confidently identify a wider range of SVs with greater precision than ever before.
Moreover, Sniffles2 addresses a critical need in genomic research by enabling the seamless transition from family-level to population-level SV calling. By producing fully genotyped Variant Call Format (VCF) files, researchers can now analyze SVs across diverse populations with ease.
The true test of Sniffles2’s capabilities came in its application to real-world scenarios. In one instance, researchers accurately identified causative SVs around the MECP2 gene, including highly complex alleles with three overlapping SVs, across 11 probands. This breakthrough opens doors for understanding the genetic basis of diseases and developing targeted therapies.
Furthermore, Sniffles2 demonstrated its prowess in detecting mosaic SVs, which are genetic alterations present in only a subset of cells within an individual. By analyzing bulk long-read data from brain tissue samples, researchers identified multiple mosaic SVs in a patient with multiple system atrophy. These SVs exhibited remarkable diversity within the cingulate cortex, impacting genes involved in neuron function and repetitive elements.
In conclusion, Sniffles2 represents a significant leap forward in the field of genomics, offering researchers a powerful tool for unraveling the mysteries hidden within the genome. With its unparalleled accuracy, efficiency, and versatility, Sniffles2 promises to accelerate discoveries in genetic research and pave the way for personalized medicine tailored to individual genomic profiles. As we continue to harness the power of long-read sequencing technology, the future of genomic medicine shines brighter than ever before.
Availability – Source code for Sniffles2 is available at https://github.com/fritzsedlazeck/Sniffles and https://doi.org/10.5281/zenodo.8121996.
Smolka M, Paulin LF, Grochowski CM, Horner DW, Mahmoud M, Behera S, Kalef-Ezra E, Gandhi M, Hong K, Pehlivan D, Scholz SW, Carvalho CMB, Proukakis C, Sedlazeck FJ. (2024) Detection of mosaic and population-level structural variants with Sniffles2. Nat Biotechnol [Epub ahead of print]. [article]


