

Long-read RNA sequencing (lrRNA-seq) produces detailed information about full-length transcripts, including novel and sample-specific isoforms. Furthermore, there is an opportunity to call variants directly from lrRNA-seq data. However, most state-of-the-art variant callers have been developed for genomic DNA.
Researchers from the Swiss Institute of Bioinformatics set out to perform a mini-benchmark on GATK, DeepVariant, Clair3, and NanoCaller primarily on PacBio Iso-Seq, data, but also on Nanopore and Illumina RNA-seq data. The researchers propose a pipeline to process spliced-alignment files, making them suitable for variant calling with DNA-based callers. With such manipulations, high calling performance can be achieved using DeepVariant on Iso-seq data.
Alignment file transformation for optimized calling of genetic variants from lrRNA-seq data and variant calling performance across the best pipelines on PacBio Iso-Seq reference datasets
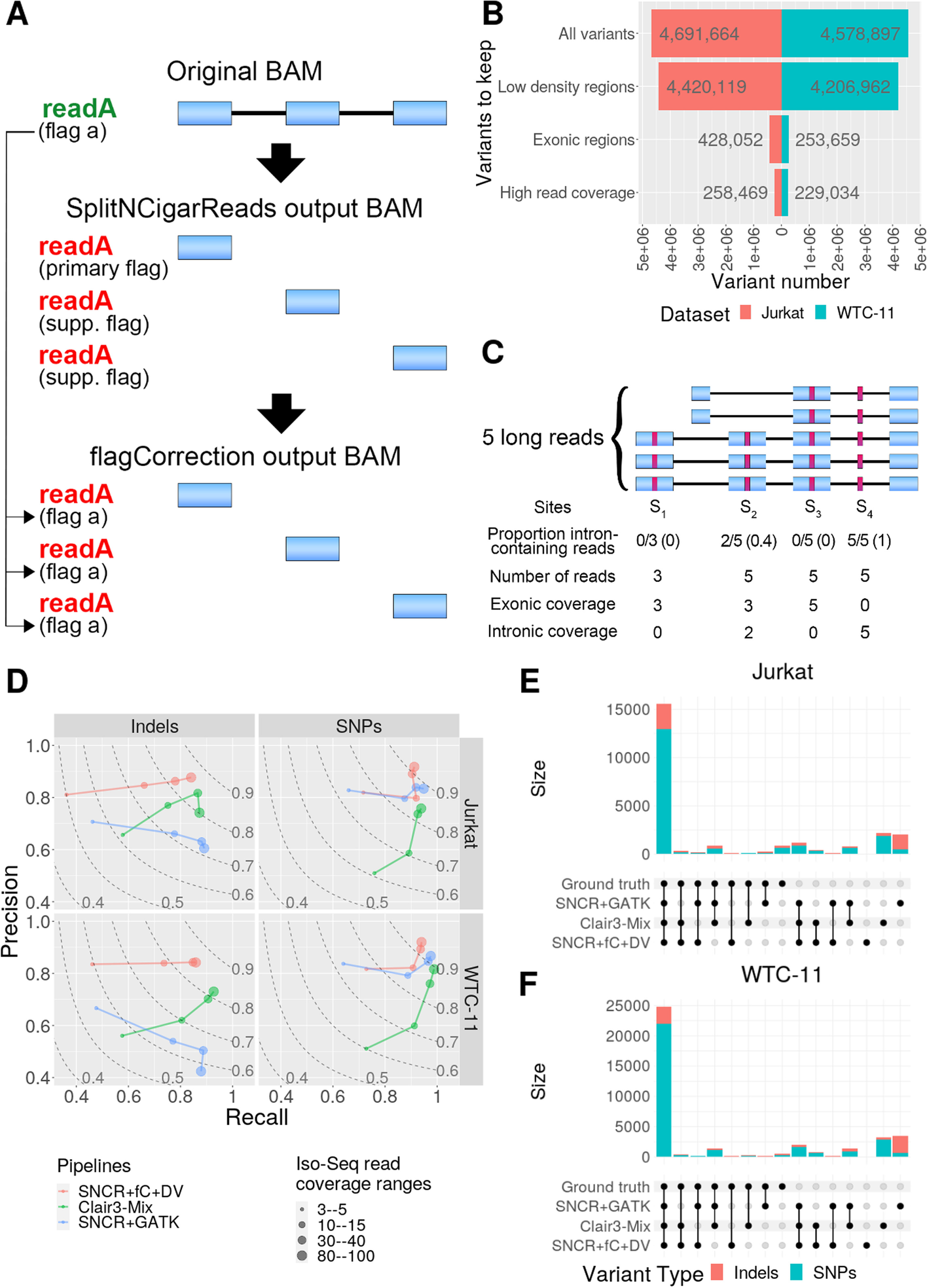
A Alignment file (BAM) transformations to make spliced lrRNA-seq alignments suitable for variant calling. First, GATK’s SNCR function is used to split the reads at Ns in their cigar string, such that exons become distinct reads. Second, the flagCorrection function attributes the flag of the original read to all corresponding fragment reads. B The number of genetic variants kept in the ground-truth (Illumina DNA-seq) variant call format (VCF) files (for Jurkat and WTC-11 datasets) after filtering; y-axis refers to variant sites that are successively retained, as follows: All variants, all sites in the VCF files; Low-density regions, sites residing in regions such that there is a maximum of 3 variants in a 201-bp window; Exonic regions, sites where the Iso-Seq coverage is at least 1; High read coverage, sites where the short-read coverage is at least 20 and 72 for Jurkat and WTC-11, respectively. C Schematic with proportion of intron-containing reads (N-cigar reads) at four variant sites (red boxes). D Precision-recall curves; point sizes indicate the filtering ranges for read coverage; dashed lines represent F1-scores. “Clair3-mix” denotes using Clair3 to call SNPs and SNCR + flagCorrection + Clair3 to call indels. SNCR-SplitNCigarReads; fC-flagCorrection; DV-DeepVariant. Additional file 1: Table S1 gives the number of covered true variants in each interval range. E, F UpSet plots show the intersection of variants called by the pipelines with the ground truth for Jurkat (E) and WTC-11 (F) datasets; sites shown here were filtered according to a minimum Iso-Seq read coverage of 20
Availability – https://github.com/vladimirsouza/lrRNAseqVariantCalling
de Souza VBC, Jordan BT, Tseng E, Nelson EA, Hirschi KK, Sheynkman G, Robinson MD. (2023) Transformation of alignment files improves performance of variant callers for long-read RNA sequencing data. Genome Biol 24(1):91. [article]


