

Transcriptome profiling is a mainstay of translational cancer research and is increasingly finding its way into precision oncology. While bulk RNA sequencing (RNA-seq) is widely available, high investment costs and long data return time are limiting factors for clinical applications. Researchers at the German Cancer Research Center (DKFZ) investigated a portable nanopore long-read sequencing device (MinION, Oxford Nanopore Technologies) for transcriptome profiling of tumors. In particular, they investigated the impact of lower coverage than that of larger sequencing devices by comparing shallow nanopore RNA-seq data with short-read RNA-seq data generated using reversible dye terminator technology (Illumina) for ten samples representing four cancer types. Coupled with ShaNTi (Shallow Nanopore sequencing for Transcriptomics), a newly developed data processing pipeline, a turnaround time of five days was achieved. The correlation of normalized gene-level counts between nanopore and Illumina RNA-seq was high for MinION but not for very low-throughput Flongle flow cells (r = 0.89 and r = 0.24, respectively). A cost-saving approach based on multiplexing of four samples per MinION flow cell maintained a high correlation with Illumina data (r = 0.56–0.86). In addition, the researchers compared the utility of nanopore and Illumina RNA-seq data for analysis tools commonly applied in translational oncology: (1) Shallow nanopore and Illumina RNA-seq were equally useful for inferring signaling pathway activities with PROGENy. (2) Highly expressed genes encoding kinases targeted by clinically approved small-molecule inhibitors were reliably identified by shallow nanopore RNA-seq. (3) In tumor microenvironment composition analysis, quanTIseq performed better than CIBERSORT, likely due to higher average expression of the gene set used for deconvolution. (4) Shallow nanopore RNA-seq was successfully applied to detect fusion genes using the JAFFAL pipeline. These findings suggest that shallow nanopore RNA-seq enables rapid and biologically meaningful transcriptome profiling of tumors, and warrants further exploration in precision cancer medicine studies.
Comparison of shallow nanopore and Illumina RNA-seq
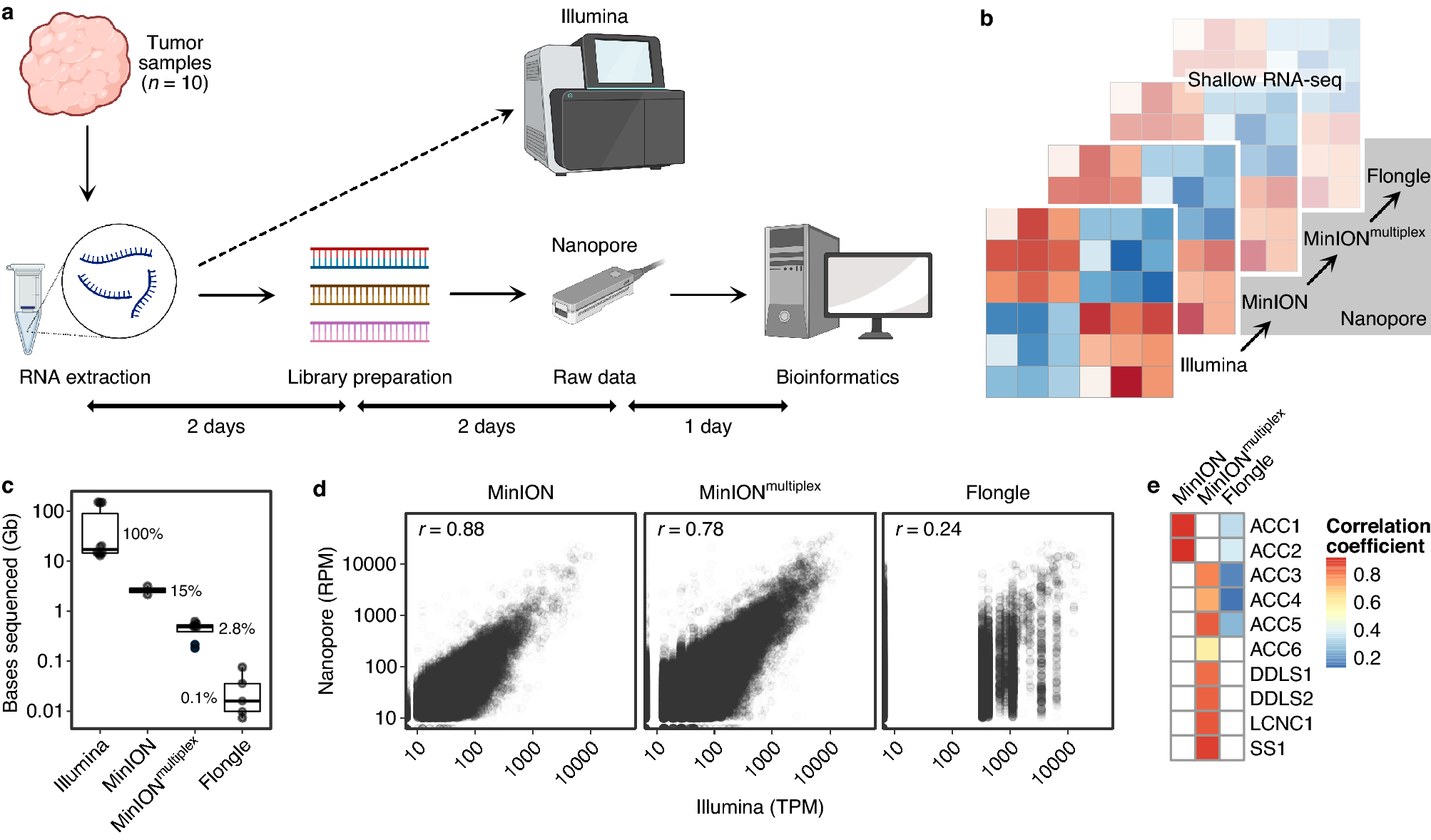
(a) Overview of sample and data processing for nanopore RNA-seq. Created with BioRender.com. (b) Schematic of the different sequencing depths used to perform nanopore RNA-seq (MinION flow cell with one sample, MinION flow cell with four samples [MinIONmultiplex], Flongle flow cell with one sample). For illustration, transcriptomic profiling is depicted by a heatmap with increasing transparency representing decreasing sequencing depth. (c) Comparison of sequencing depth, i.e., bases sequenced, between technologies. Nanopore RNA-seq covered a median of 0.1–15% of the bases sequenced with Illumina RNA-seq. (d) Correlation of normalized gene-level counts obtained by nanopore (RPM values) and Illumina (TPM values) RNA-seq across all samples and protein-coding genes (Spearman rank correlation coefficient). Each dot represents a gene. (e) Correlation of normalized gene-level counts obtained by nanopore and Illumina RNA-seq for individual samples (Spearman rank correlation coefficient).
Availability – Source code for the ShaNTi pipeline is available at https://github.com/cihanerkut/shanti. Source code for the RNA-Seq workflow of the ODCF is available at https://github.com/DKFZ-ODCF/RNAseqWorkflow.
Mock A, Braun M, Scholl C, Fröhling S, Erkut C. (2023) Transcriptome profiling for precision cancer medicine using shallow nanopore cDNA sequencing. Sci Rep 13(1):2378. [article]


