

Since many single-cell RNA-seq (scRNA-seq) data are obtained after cell sorting, such as when investigating immune cells, tracking cellular landscape by integrating single-cell data with spatial transcriptomic data is limited due to cell type and cell composition mismatch between the two datasets. Researchers at the Seoul National University have developed a method, spSeudoMap, which utilizes sorted scRNA-seq data to create virtual cell mixtures that closely mimic the gene expression of spatial data and trains a domain adaptation model for predicting spatial cell compositions. The method was applied in brain and breast cancer tissues and accurately predicted the topography of cell subpopulations. spSeudoMap may help clarify the roles of a few, but crucial cell types.
Mapping cell subpopulations to the spatial transcriptomic data with spSeudoMap
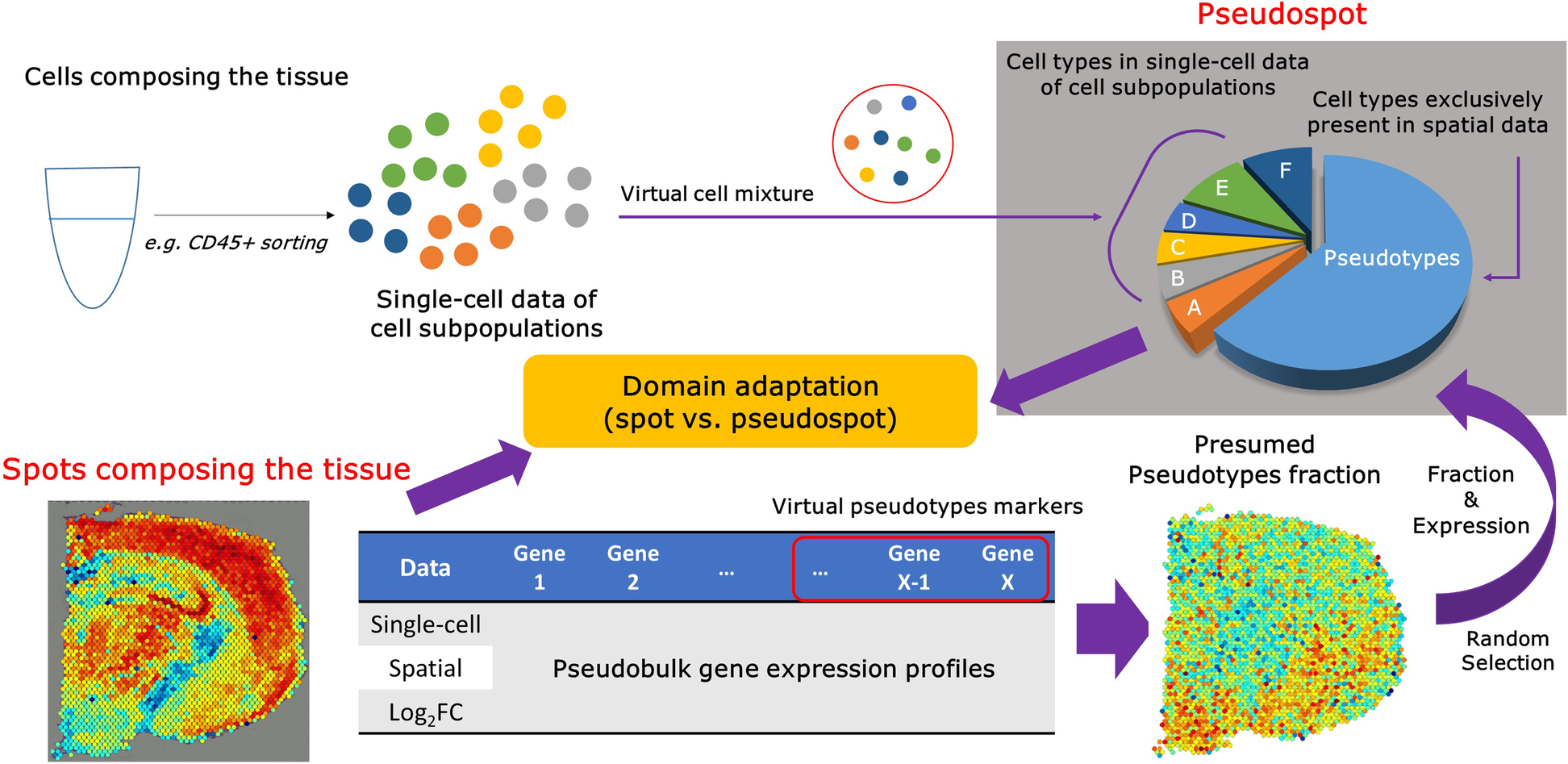
The cell types of the single-cell transcriptomic data acquired from cell sorting experiments can be spatially mapped to the tissue using spSeudoMap. The single-cell data of cell subpopulations are composed of sorted cells from the tissue, and the cell types cover only part of those in the spatial transcriptomics data. To create the reference dataset that mimics the spatial data, virtual cell mixtures, pseudospots, are defined in which all cell types from the tissue are included. First, the cell types exclusively present in the spatial data are aggregated and named pseudotypes. The virtual markers for the pseudotypes are selected from the top genes highly expressed in spatial pseudobulk compared to single-cell pseudobulk data. Then, the pseudotype fraction in the spatial spots is estimated from the module scores (sc.tl.score_genes in Scanpy) of the top 20 pseudotype markers. The fraction and gene expression of the pseudotypes are assigned based on the presumed pseudotype fraction and expression of a randomly selected spatial spot. Lastly, the target type proportion of the pseudospot, explained by cell types in the single-cell data, is filled with the randomly sampled cells from the single-cell data of cell subpopulations. Finally, the pseudospot is considered a reference dataset for the domain adaptation method CellDART
Availability – Python source code and R wrap function for spSeudoMap is uploaded on https://github.com/bsungwoo/spSeudoMap.
Bae S, Choi H, Lee DS. (2023) spSeudoMap: cell type mapping of spatial transcriptomics using unmatched single-cell RNA-seq data. Genome Med 15(1):19. [article]


