

RNA plays a crucial role in all living cells, acting as a messenger that carries genetic instructions from DNA to make proteins. Scientists are keen to understand RNA in more detail, especially the chemical modifications it undergoes, as these can affect its function. One advanced method used to study these modifications is called nucleotide conversion RNA sequencing. This technique involves making specific changes to the RNA molecules and then sequencing them to see where these changes occur. However, this process can introduce errors or “mismatches” when comparing the new RNA sequences to reference genomes. Understanding these errors is essential for accurate data interpretation.
The Problem with Mapping Mismatched Reads
When scientists use nucleotide conversion RNA sequencing, they often encounter mismatches in the data. These mismatches can create biases when mapping the RNA sequences back to reference genomes (the complete set of genetic information for a species). If these biases are not well understood or corrected, the conclusions drawn from the data can be misleading. This is where the new tool, splice_sim, comes into play.
What is splice_sim?
Splice_sim is a powerful and innovative pipeline developed by researchers at the Vienna Biocenter Campus and designed to simulate and evaluate RNA sequencing data. Here’s a breakdown of what it does:
- User-Defined Nucleotide Conversions: Splice_sim allows users to introduce specific nucleotide conversions (changes in the RNA sequence) at set frequencies. This helps in mimicking the modifications that naturally occur in RNA molecules.
- Creating Mixture Models: It generates mixture models of both converted (modified) and unconverted (original) RNA reads. This means splice_sim can create a realistic mix of RNA sequences as they would appear in an actual experiment.
- Calculating Mapping Accuracies: The tool then calculates how accurately these modified RNA sequences can be mapped back to the reference genomes. It assesses the performance of various state-of-the-art RNA sequencing mappers, which are software tools used to align RNA sequences to the genome.
Analysis workflow and NC mapping accuracies for simulated mouse metabolic labeling data
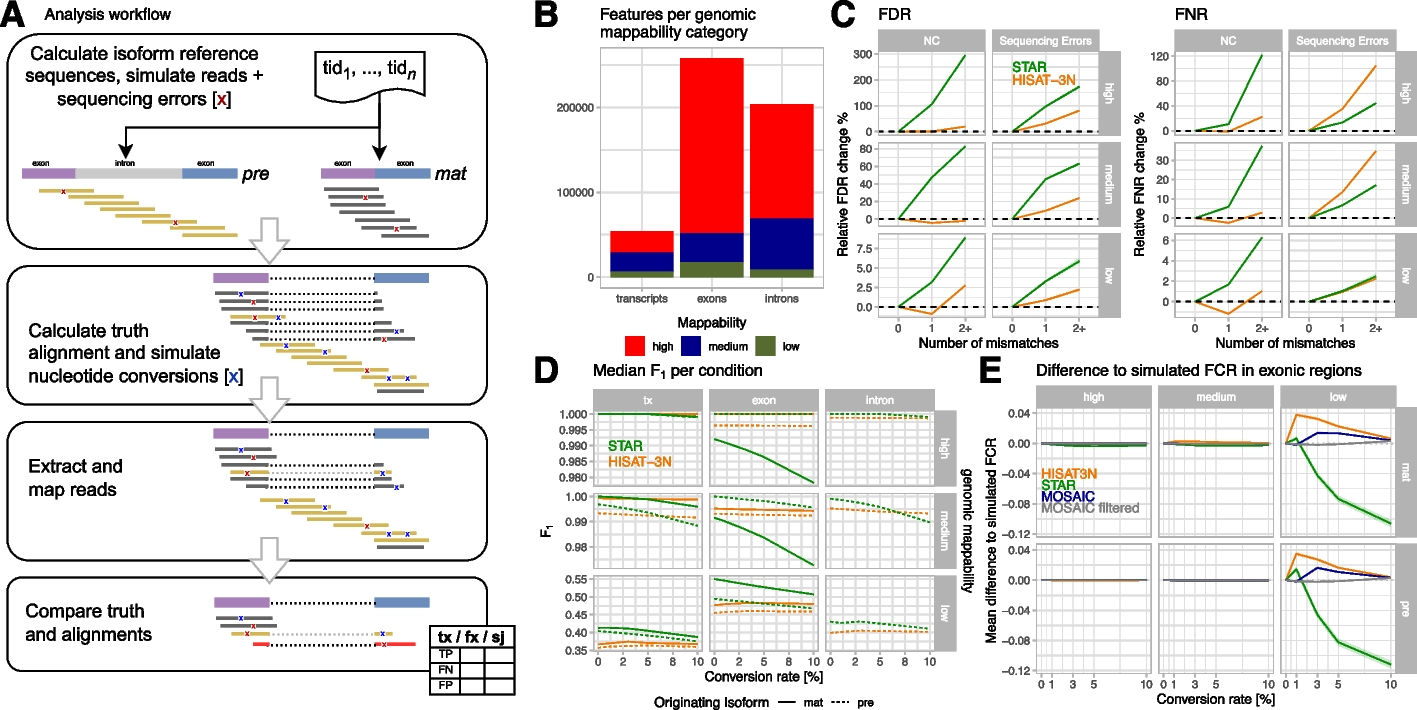
A Analysis workflow overview: briefly, we simulated short reads with realistic sequencing error (red X) for premature and mature isoforms, calculated truth alignments, and injected nucleotide conversions with configured conversion rates. Simulated reads were mapped by the evaluated read mappers and resulting alignments were compared to the simulated data. Finally, grouped count tables with true positive (TP), false positive (FP), and false negative (FN) counts per annotation of interest (tx: transcripts, fx: exons + introns, sj: splice junctions) were created and analyzed. B Numbers of analyzed m_big annotations with high (> 0.9), medium, and low (< 0.2) mean genome mappability. C Changes of false discovery (FDR = FP/(TP + FP) and false negative (FNR = FN/(TP + FN)) rates by number of mismatches per read compared to reads without mismatches, stratified by mappability and type of mismatch (either simulated NC or random sequencing errors). The plots show median FDR/FNR and interquartile regions (shaded areas) across three m_big replicates for STAR (green) and HISAT-3N (orange) alignments. This analysis included ~ 12B reads originating from premature isoforms and their classification (TP, FP, FN) with respect to whole-transcript annotations. D Median F1 measure per mapper and originating isoform (pre: premature, mat: mature) for different genomic annotations (tx: whole transcript), stratified by mappability. E Mean difference to simulated, exonic FCR (fraction of converted reads) per mapper and for a “mosaic” approach where the mapper with the smallest difference to the simulated value was chosen. The mosaic approach reduces differences to simulated values and when removing exons where none of the two mappers showed good results, reconstruction is nearly perfect (“mosaic filtered,” see main text).
Why is splice_sim Important?
By using splice_sim, researchers can simulate RNA sequencing under realistic experimental conditions. For example, it can replicate scenarios involving metabolic RNA labeling (a method to track new RNA synthesis) and RNA bisulfite sequencing (a technique to study RNA modifications). Through these simulations, splice_sim measures the mapping accuracies of the current best RNA mappers for both mouse and human transcripts.
Preventing Data Interpretation Biases
One of the key benefits of splice_sim is its ability to identify and suggest strategies to prevent biases in data interpretation. By understanding where and why mismatches occur, researchers can improve the accuracy of their RNA sequencing studies. This leads to more reliable results, which are crucial for advancing our knowledge of RNA biology and its implications in health and disease.
Conclusion
In summary, splice_sim is a valuable tool for researchers studying RNA modifications. It helps simulate realistic RNA sequencing conditions, evaluates mapping accuracies, and provides insights into preventing biases in data interpretation. This advancement represents a significant step forward in the field of RNA research, enabling scientists to obtain more accurate and meaningful data from their studies.
Popitsch N, Neumann T, von Haeseler A, Ameres SL. (2024) Splice_sim: a nucleotide conversion-enabled RNA-seq simulation and evaluation framework. Genome Biol 25(1):166. [article]


