

Unsupervised clustering of single-cell RNA sequencing data (scRNA-seq) is important because it allows us to identify putative cell types. However, the large number of cells (up to millions), the high-dimensionality of the data (tens of thousands of genes), and the high dropout rates all present substantial challenges in single-cell analysis.
Researchers from the University of Nevada, Reno have developed a new method, named single-cell Clustering using Autoencoder and Network fusion (scCAN), that can overcome these challenges to accurately segregate different cell types in large and sparse scRNA-seq data. In an extensive analysis using 28 real scRNA-seq datasets (more than three million cells) and 243 simulated datasets, the researchers validate that scCAN: (1) correctly estimates the number of true cell types, (2) accurately segregates cells of different types, (3) is robust against dropouts, and (4) is fast and memory efficient. We also compare scCAN with CIDR, SEURAT3, Monocle3, SHARP, and SCANPY. scCAN outperforms these state-of-the-art methods in terms of both accuracy and scalability.
The overall analysis pipeline of scCAN consists of three modules
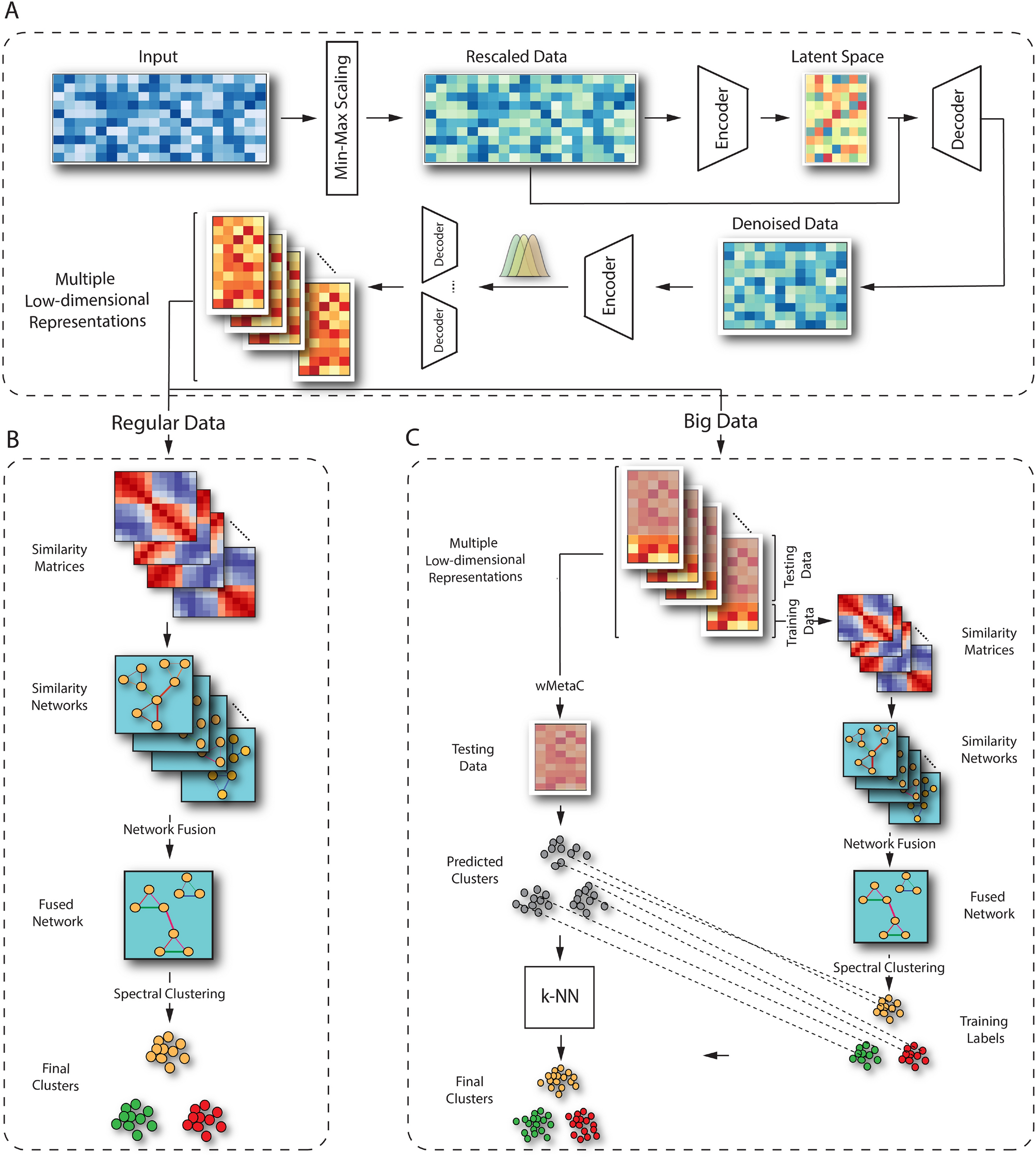
In the first module (A), we perform data normalization, gene filtering, and latent variables generation using two autoencoders. In the second module (B), we adopt the network fusion-based clustering method to segregate cell types for small data. The third module (C) aims at clustering big data using a combination of the network fusion approach and K nearest neighbors (k-NN) algorithm.
Availability – The scCAN package is available at https://cran.r-project.org/package=scCAN. Data and R scripts are available at http://sccan.tinnguyen-lab.com/
Tran B, Tran D, Nguyen H, Ro S, Nguyen T. scCAN: single-cell clustering using autoencoder and network fusion. Sci Rep 12(1):10267. [article]


