

Characterization of microbial activity is essential to the understanding of the basic biology of microbial communities, as the function of a microbiome is defined by its biochemically active (“viable”) community members. Current sequence-based technologies can rarely differentiate microbial activity, due to their inability to distinguish live and dead sourced DNA. As a result, our understanding of microbial community structures and the potential mechanisms of transmission between humans and our surrounding environments remains incomplete. As a potential solution, 16S rRNA transcript-based amplicon sequencing (16S-RNA-seq) has been proposed as a reliable methodology to characterize the active components of a microbiome, but its efficacy has not been evaluated systematically. Researchers at the Harvard T.H. Chan School of Public Health present their work to benchmark RNA-based amplicon sequencing for activity assessment in synthetic and environmentally sourced microbial communities.
In synthetic mixtures of living and heat-killed Escherichia coli and Streptococcus sanguinis, 16S-RNA-seq successfully reconstructed the active compositions of the communities. However, in the realistic environmental samples, no significant compositional differences were observed in RNA (“actively transcribed — active”) vs. DNA (“whole” communities) spiked with E. coli controls, suggesting that this methodology is not appropriate for activity assessment in complex communities. The results were slightly different when validated in environmental samples of similar origins (i.e., from Boston subway systems), where samples were differentiated both by environment type as well as by library type, though compositional dissimilarities between DNA and RNA samples remained low (Bray–Curtis distance median: 0.34–0.49). To improve the interpretation of 16S-RNA-seq results, we compared our results with previous studies and found that 16S-RNA-seq suggests taxon-wise viability trends (i.e., specific taxa are universally more or less likely to be viable compared to others) in samples of similar origins.
16S-RNA-seq indicated subtle differentiation between DNA and RNA libraries in samples from the Boston (MBTA) subway system
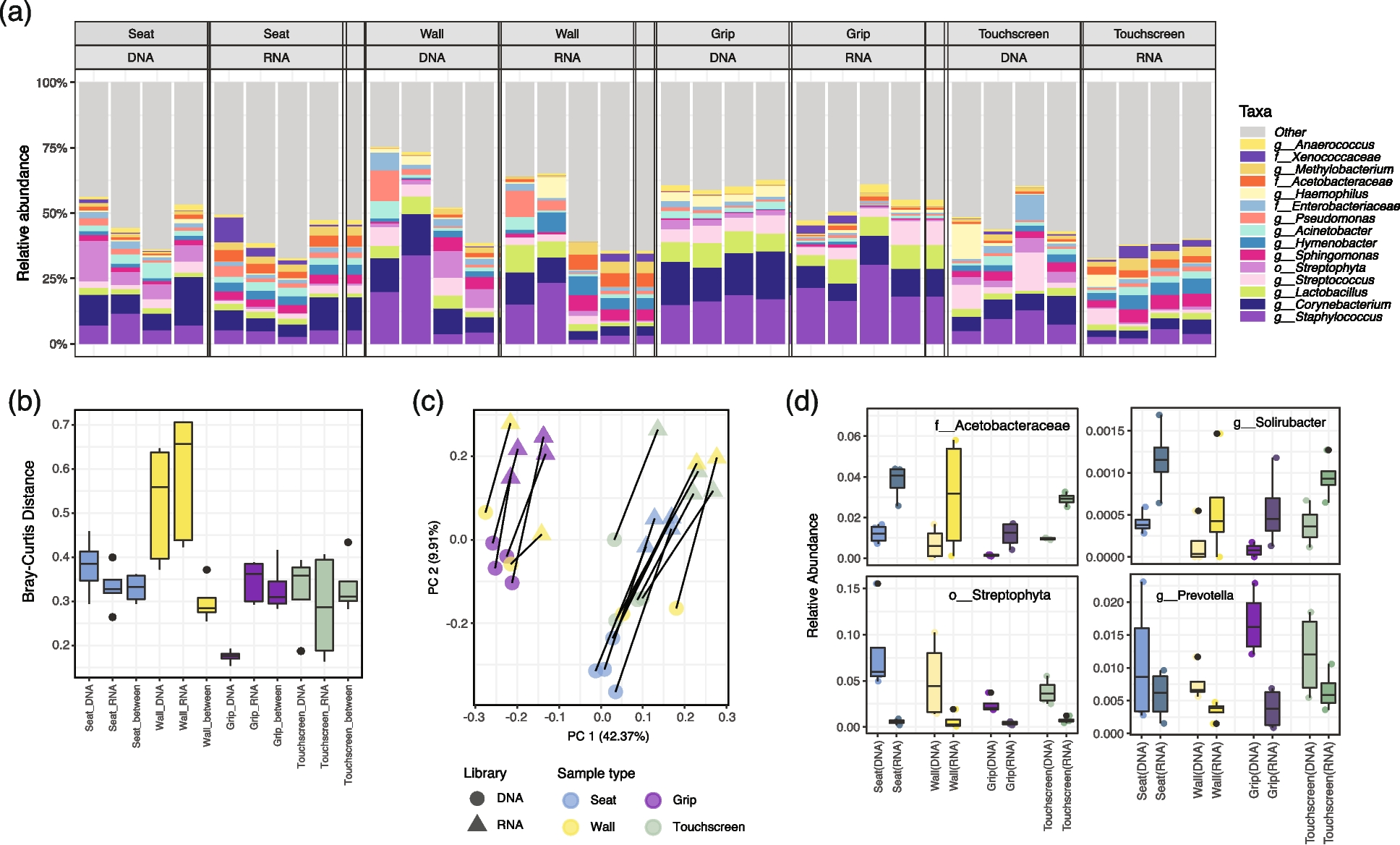
a Relative abundances of the 15 taxa with the highest means across four sample types in DNA and RNA libraries indicated overall similar taxonomic compositions. Each column represents a biological replicate. b Bray–Curtis distance distributions between MBTA samples within/between DNA and RNA libraries. c Principal coordinate analysis (PCoA) of MBTA samples using Bray–Curtis distances. Sample type and library type are both drivers of overall community composition. d Four differentially abundant taxa that consistently enriched or depleted in RNA libraries across sample types. The Acetobacteraceae family and Solirubacter genus were consistently enriched in RNA libraries, while the Streptophyta order and the human commensal Prevotella genus were under quantified in RNA libraries (mixed-effects linear models, q-value = 0.002, 0.003, 0.002, and 0.012, respectively)
This study provides a comprehensive evaluation of 16S-RNA-seq for viability assessment in synthetic and complex microbial communities. The results found that while 16S-RNA-seq was able to semi-quantify microbial viability in relatively simple communities, it only suggests a taxon-dependent “relative” viability in realistic communities.
Wang Y, Thompson KN, Yan Y, Short MI, Zhang Y, Franzosa EA, Shen J, Hartmann EM, Huttenhower C. (2023) RNA-based amplicon sequencing is ineffective in measuring metabolic activity in environmental microbial communities. Microbiome 11(1):131. [article]


