

Accurate and comprehensive annotation of transcript sequences is essential for transcript quantification and differential gene and transcript expression analysis. Single-molecule long-read sequencing technologies provide improved integrity of transcript structures including alternative splicing, and transcription start and polyadenylation sites. However, accuracy is significantly affected by sequencing errors, mRNA degradation, or incomplete cDNA synthesis.
Researchers at the James Hutton Institute present a new and comprehensive Arabidopsis thaliana Reference Transcript Dataset 3 (AtRTD3). AtRTD3 contains over 169,000 transcripts-twice that of the best current Arabidopsis transcriptome and including over 1500 novel genes. Seventy-eight percent of transcripts are from Iso-seq with accurately defined splice junctions and transcription start and end sites. The researchers developed novel methods to determine splice junctions and transcription start and end sites accurately. Mismatch profiles around splice junctions provide a powerful feature to distinguish correct splice junctions and remove false splice junctions. Stratified approaches identify high-confidence transcription start and end sites and remove fragmentary transcripts due to degradation. AtRTD3 is a major improvement over existing transcriptomes as demonstrated by analysis of an Arabidopsis cold response RNA-seq time-series. AtRTD3 provides higher resolution of transcript expression profiling and identifies cold-induced differential transcription start and polyadenylation site usage.
Workflow of analysis of PacBio Iso-sequencing
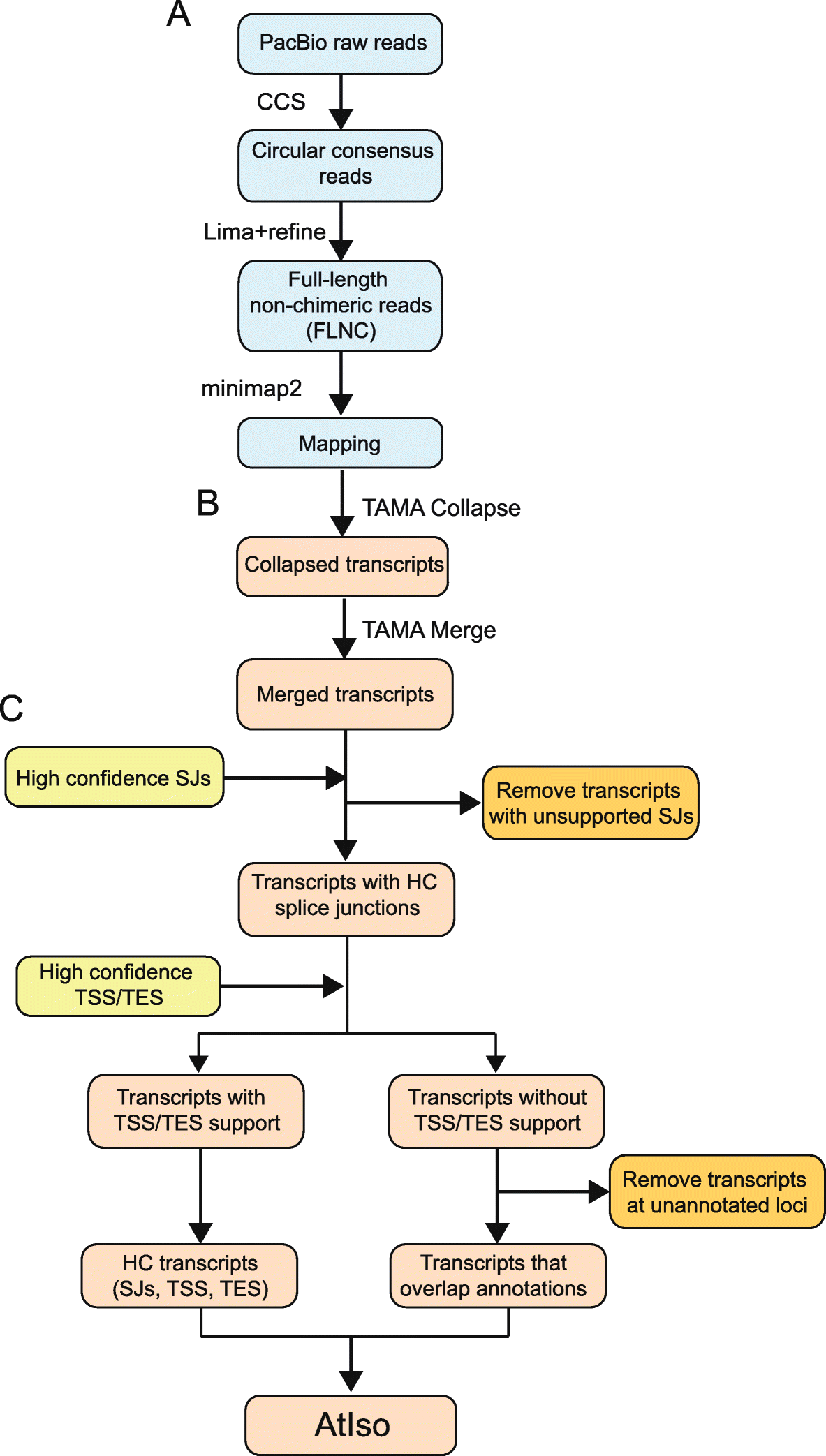
A Raw reads are analyzed using the PacBio Iso-seq 3 pipeline to generate FLNCs which are mapped to the genome (blue boxes). B Mapped FLNCs are collapsed and merged using TAMA to generate transcripts (pink boxes). C Transcripts are quality controlled using datasets of high-confidence (HC) splice junctions (SJs) and transcript start and end sites (TSS/TES). Transcripts with unsupported splice junctions where reads contain mismatches within ±10 nt of an SJ are removed. Transcripts with both high-confidence TSS and TES (determined by binomial probability for highly expressed genes and by end support with > 2 reads for low expressed genes) are retained as HC transcripts. The remaining transcripts which have partial or no TSS and/or TES support were removed unless they overlapped with annotated gene loci. These transcripts, from genes with low coverage by Iso-seq, were combined with the HC transcripts to form AtIso (Arabidopsis Iso-seq based transcriptome)
AtRTD3 is the most comprehensive Arabidopsis transcriptome currently. It improves the precision of differential gene and transcript expression, differential alternative splicing, and transcription start/end site usage analysis from RNA-seq data. The novel methods for identifying accurate splice junctions and transcription start/end sites are widely applicable and will improve single-molecule sequencing analysis from any species.
Availability – The AtRTD3 annotations are available in fasta, bed, and gtf format at https://ics.hutton.ac.uk/atRTD/RTD3/
Zhang R, Kuo R, Coulter M, Calixto CPG, Entizne JC, Guo W, Marquez Y, Milne L, Riegler S, Matsui A, Tanaka M, Harvey S, Gao Y, Wießner-Kroh T, Paniagua A, Crespi M, Denby K, Hur AB, Huq E, Jantsch M, Jarmolowski A, Koester T, Laubinger S, Li QQ, Gu L, Seki M, Staiger D, Sunkar R, Szweykowska-Kulinska Z, Tu SL, Wachter A, Waugh R, Xiong L, Zhang XN, Conesa A, Reddy ASN, Barta A, Kalyna M, Brown JWS. (2022) A high-resolution single-molecule sequencing-based Arabidopsis transcriptome using novel methods of Iso-seq analysis. Genome Biol 23(1):149. [article]


