

Advancements in single-cell genomics have revolutionized our understanding of cell diversity and function in complex biological systems. Among the most widely used technologies for single-cell analysis are chromatin accessibility profiling and transcriptome sequencing. In a recent development, researchers from the Zhejiang University School of Medicine have introduced Microwell-seq3, a novel platform designed to facilitate high-throughput and sensitive profiling of chromatin accessibility and transcriptomes at the single-nucleus level. This groundbreaking technology promises to significantly enhance our ability to unravel the intricacies of cellular heterogeneity and gene regulation across diverse tissues and diseases.
Microwell-seq3 employs a preindexing strategy and a unique penetrable chip-in-a-tube design, enabling seamless loading of single nuclei and efficient DNA amplification without the need for specialized equipment. This streamlined approach allows researchers to profile chromatin accessibility or full-length transcriptomes with high sensitivity and throughput, making it a versatile tool for various research applications.
Overview of the microwell-seq3 workflow
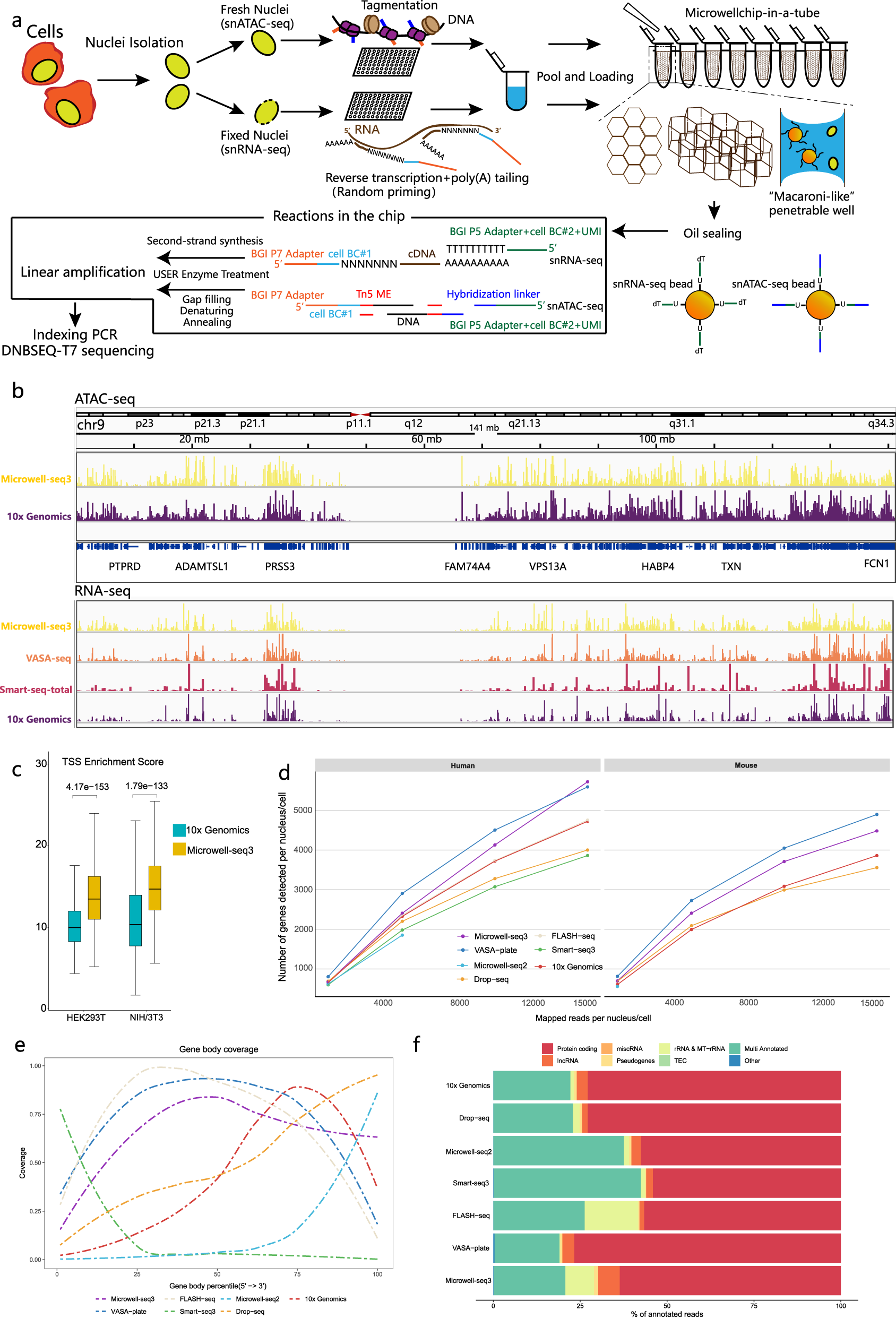
a Schematic view of the Microwell-seq3 workflow. Single nuclei are extracted from fresh or frozen tissues. DNA or RNA in nuclei is tagged with the first part of the cell barcode (BC#1) in multiple 96-well plates. Labeled nuclei and barcoded magnetic beads are pooled and evenly loaded into Microwell chips. Oligos are released from the beads and linear preamplification is performed in the chips. Preamplified DNA or RNA fragments are collected from the chips for final library preparation and sequencing. b Representative University of California Santa Cruz (UCSC) Genome Browser view of ATAC-seq and RNA-seq signal tracks (HEK293T cells) from Microwell-seq3, 10X Genomics, VASA-seq and Smart-seq-total data. c Distribution of TSS enrichment scores from Microwell-seq3 and 10X Genomics ATAC-seq data in the indicated cell lines, reads (fragments) are down sampled to 2000 fragments per nucleus. The statistical test used is a two-sided Student’s t-test. d Number of the detected annotated genes in human and mouse cell lines (HEK293T, mouse NIH/3T3 and embryonic stem cells (mESCs)) in each method is plotted against the number of unique mapped reads per cell in different down-sampling thresholds. All the reads are remapped with the same pipeline after adapter filtering and trimming. Data of mESCs were generated by VASA-seq. e Comparison of mean ± standard deviation (SD) gene body coverage in protein-coding genes in cell lines across the different methods. Microwell-seq3, FLASH-seq and VASA-seq show even coverage across the length of the genes. Other methods show bias toward the 5′ and 3′ ends of transcripts, respectively. Reads in each method are trimmed to 500,000 per sample. f Proportions of the reads mapped to all annotated genes for each biotype in cell lines across the different methods. Microwell-seq3 detects proportionally higher levels of lncRNAs. Reads in each method are trimmed to 5000 per cell.
In a remarkable demonstration of its capabilities, the researchers utilized Microwell-seq3 to analyze chromatin accessibility in over 200,000 single nuclei and the full-length transcriptome in approximately 50,000 nuclei from multiple adult mouse tissues. By integrating cell type-specific regulatory elements with total RNA expression data, they uncovered comprehensive cell type heterogeneity in the brain, providing valuable insights into the molecular mechanisms underlying neural diversity and function.
Moreover, the gene regulatory networks derived from chromatin accessibility profiling offered an improved model of cell type communication, shedding light on the complex interactions between different cell populations within tissues. Importantly, Microwell-seq3 demonstrated its utility in disease research by accurately identifying malignant cells and their specific regulons in spontaneous lung tumors of aged mice. This capability holds significant promise for studying tumor heterogeneity and identifying potential therapeutic targets.
Microwell-seq3 represents a significant advancement in the field of single-cell genomics, offering researchers a powerful tool to explore cell diversity, gene regulation, and disease pathology with unprecedented sensitivity and throughput. By enabling comprehensive profiling of chromatin accessibility and transcriptomes at the single-nucleus level, this innovative platform holds great potential for advancing our understanding of complex biological systems and accelerating discoveries in various areas of research, from neuroscience to cancer biology. As researchers continue to harness the capabilities of Microwell-seq3, we can anticipate exciting new insights into the fundamental principles governing cellular function and dysfunction.
Ye F, Zhang S, Fu Y et al. (2024) Fast and flexible profiling of chromatin accessibility and total RNA expression in single nuclei using Microwell-seq3. Cell Discov [Epub ahead of print]. [article]


