

Understanding the function of rare non-coding variants has long been a daunting task. These elusive variants, residing in regions of the genome that do not code for proteins, play a crucial role in regulating gene expression post-transcriptionally. Now, a groundbreaking study by researchers at the University of California, Los Angeles utilizing a screening method called MapUTR has shed light on the function of these rare variants, particularly those found in the 3′ untranslated regions (UTRs) of messenger RNA (mRNA).
MapUTR captures functional 3′ UTR variants in known functional motifs
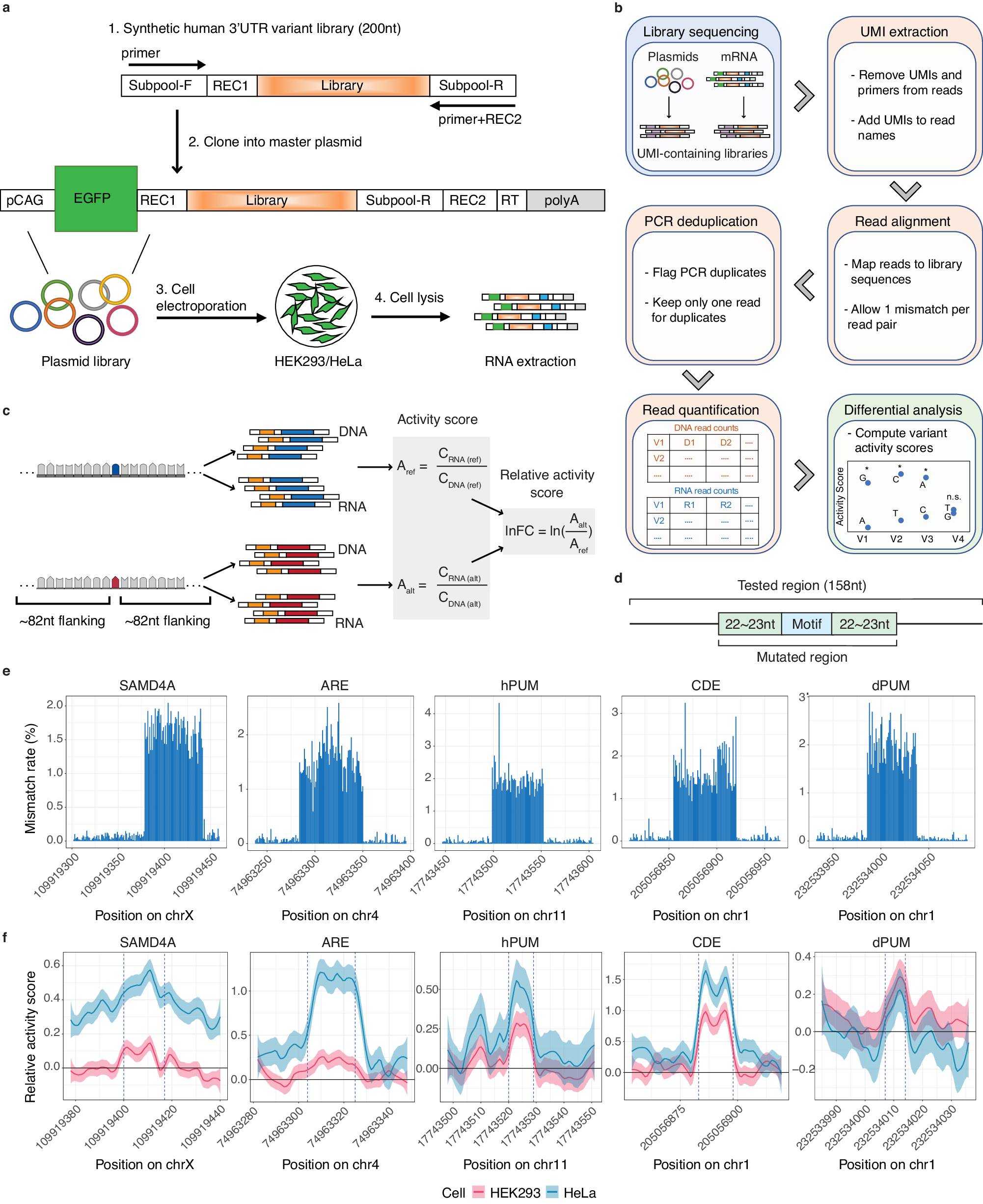
a Experimental Design of MapUTR. Subpool-F and Subpool-R represent the 15-mer primer sequences to amplify the oligo pool. REC1 and REC2 represent restriction enzyme sites. pCAG, the CMV early enhancer/chicken beta actin (CAG) promoter. EGFP, eGFP gene. Library, oligo sequences containing the mutations/motifs. RT, sequences for gene-specific reverse transcription. polyA, polyA signals. See also Supplementary Fig. 2. b Computational workflow. c Design of oligos containing rare variants from gnomAD and quantification of variant effects. CDNA (ref) represents DNA counts for the reference allele, CRNA (ref) represents RNA counts for the reference allele, CDNA (alt) represents DNA counts for the alternative allele, CRNA (alt) represents RNA counts for the alternative allele, Aref represents the activity score of the reference allele, Aalt represents the activity score of the alternative allele, and lnFC represents the relative activity score of the alternative allele compared to the reference allele. Created with BioRender.com. d Diagram of oligos with random mutations in the motif and its flanking regions (22–23 nt). e Mismatch rate (%) per position along the length of DNA sequences harboring known motifs: SAMD4A, sterile alpha motif domain containing 4A motif (in gene CHRDL1), ARE, AU-rich element (in gene CXCL2), hPUM, human pumilio motif (in gene MYOD1), CDE, constitutive decay element (in gene RBBP5), and dPUM, Drosophila pumilio motif (in gene SIPA1L2). f Relative activity scores of each mutated position of known motifs in HEK293 (red) and HeLa (blue) cells. Relative activity scores are averaged across the 3 tested alternative alleles per position. The 95% confidence intervals are shown as shades. e, f Source data are provided as a Source Data file.
The study, which analyzed a vast dataset comprising 17,301 rare variants, revealed that a significant portion—approximately 24.5%—exhibited functional effects on mRNA abundance. Strikingly, many of these functional variants were located in genes associated with cancer, including those involved in critical cancer pathways. This discovery prompted further investigation into somatic mutations—those acquired during an individual’s lifetime—in cancer driver genes. Remarkably, the analysis uncovered that 33% of somatic mutations were functional, implicating them in cancer development and progression.
The functional MapUTR variants were found to be enriched in regions known as microRNA- or protein-binding sites, suggesting their involvement in regulating gene expression. Moreover, these variants may contribute to outlier gene expression patterns observed in tumors, providing valuable insights into the molecular mechanisms underlying cancer.
One of the most significant findings of the study is the introduction of a novel metric called untranslated tumor mutational burden (uTMB). This metric quantifies the amount of somatic functional MapUTR variants within a tumor and shows promise in predicting patient survival outcomes. By leveraging uTMB, researchers can gain a deeper understanding of the genetic landscape of tumors and potentially identify patients who may benefit from tailored treatment strategies.
Furthermore, the study utilized prime editing—a cutting-edge genome editing technique—to characterize three specific variants found in cancer-relevant genes (MFN2, FOSL2, and IRAK1). These experiments demonstrated the cancer-driving potential of these variants, further highlighting their significance in oncogenesis.
This study has unraveled the functional significance of tens of thousands of non-coding variants, providing crucial insights into cancer biology. By elucidating the role of these variants in regulating gene expression and driving cancer development, these researchers are paving the way for the development of novel therapeutic interventions and personalized treatment approaches for cancer patients.
Fu T, Amoah K, Chan TW, Bahn JH, Lee JH, Terrazas S, Chong R, Kosuri S, Xiao X.(2024) Massively parallel screen uncovers many rare 3′ UTR variants regulating mRNA abundance of cancer driver genes. Nat Commun 15(1):3335. [article]


