

RNA sequencing (RNA-seq) is a powerful technique used to study the transcriptome—the complete set of RNA transcripts produced by the genome. Understanding the transcriptome is crucial for insights into gene expression, cellular functions, and the underlying mechanisms of various diseases. Traditionally, RNA-seq has relied on short-read sequencing methods, which, although useful, often fall short in providing complete, accurate representations of full-length RNA molecules.
The Need for Long-Read RNA Sequencing
Long-read RNA sequencing (long-read RNA-seq) offers the promise of capturing full-length RNA transcripts, providing a more comprehensive view of the transcriptome. This is particularly important for accurately annotating eukaryotic genomes, which include complex organisms like humans and mice. Despite significant advancements, long-read sequencing technologies still face challenges, particularly in reliably identifying complete RNA transcripts from start to finish.
Introducing CapTrap-seq
To address these challenges, researchers at the Centre for Genomic Regulation have developed a new method called CapTrap-seq. This innovative technique is designed to improve the accuracy and completeness of long-read RNA sequencing. Let’s break down how CapTrap-seq works and why it’s a game-changer.
Full-length transcript annotation using CapTrap-seq and other library preparation methods
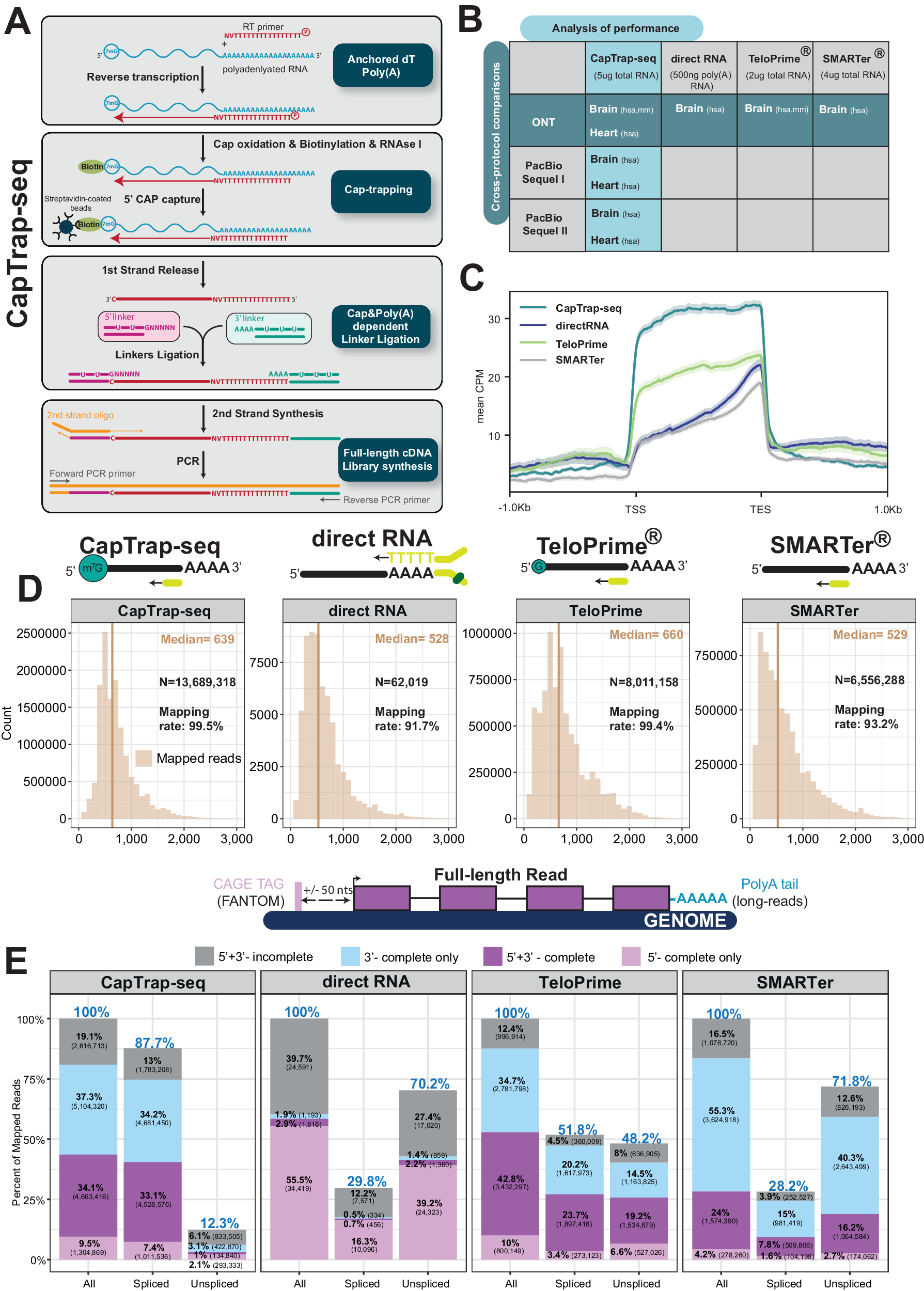
A CapTrap-seq experimental workflow. Gray boxes highlight the four main steps of full-length (FL) cDNA library construction: Anchored dT Poly(A)+, CAP-trapping, CAP and Poly(A) dependent linker ligation, and FL-cDNA library enrichment as described in the text. B Two adult human complex transcriptomic samples, brain and heart, were used to perform the cross-protocol and cross-platform comparisons to assess the quality of CapTrap-seq. The horizontal green line indicates the cross-protocol comparisons, including four different sequencing library preparation methods: CapTrap-seq, directRNA®, TeloPrime®, and SMARTer®. Whereas, the vertical blue line shows cross-platform comparison using CapTrap-seq in combination with three long-read sequencing platforms: ONT, PacBio Sequel I, and Sequel II. C Read aggregate deepTools2 profiles along the body of annotated GENCODE genes. The shaded regions indicate the 95% confidence interval. D Length distribution of mapped long-read ONT reads for each protocol. The total number of reads (N), median read length (beige vertical line), and the mapping rate are shown in the top right corner. E Detection of full-length reads among all, spliced and unspliced reads, with 5′ and 3′ termini inferred from robust (FANTOM5 phase 1 and 2 robust (n = 201,802)) CAGE clusters and poly(A) tails. Colors highlights four different categories of long-read (LR) completeness: Gray: unsupported LRs; Sky blue: 3’ supported LRs; Light pink: 5’ supported LRs; Purple: 5’ + 3’ supported LRs. The blue percentage displayed at the top of each bar indicates the ratio of a specific read type (spliced, unspliced) to the total number of reads.
How CapTrap-seq Works
- Cap-Trapping Strategy: One of the key features of CapTrap-seq is the use of a cap-trapping strategy. The 5′ end of most RNA molecules in cells has a special structure called a “cap.” This cap is crucial for the stability and proper functioning of the RNA. CapTrap-seq specifically captures these capped RNA molecules, ensuring that the transcripts are complete from the very beginning.
- Oligo(dT) Priming: In addition to cap-trapping, CapTrap-seq uses oligo(dT) priming, a technique that targets the poly(A) tail at the 3′ end of RNA molecules. By combining these two strategies, CapTrap-seq can accurately capture full-length RNA molecules from end to end.
Performance Evaluation
To validate the effectiveness of CapTrap-seq, the researchers conducted a series of tests using human and mouse tissues. They compared CapTrap-seq with other widely used RNA-seq library preparation protocols, employing two leading long-read sequencing technologies: Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio).
Benchmarking with Synthetic RNA
To further test the accuracy of CapTrap-seq, the researchers used synthetic RNA sequences with a capping strategy that mimics natural RNA. This allowed them to precisely measure how well CapTrap-seq could reconstruct full-length RNA molecules. The results were benchmarked using data from the Long-read RNA-seq Genome Annotation Assessment Project (LRGASP).
Key Findings
The benchmarks demonstrated that CapTrap-seq is a competitive and effective method for generating full-length RNA sequences. Here are the key findings:
- High Accuracy: CapTrap-seq accurately identifies full-length RNA transcripts, ensuring that researchers get a complete picture of the RNA molecules.
- Platform-Agnostic: CapTrap-seq works well with multiple sequencing technologies, making it a versatile tool for various research applications.
- Improved Annotations: By providing reliable full-length RNA sequences, CapTrap-seq helps improve the annotation of eukaryotic genomes, contributing to a better understanding of gene expression and function.
Conclusion
CapTrap-seq represents a significant advancement in the field of RNA sequencing. By combining cap-trapping with oligo(dT) priming, it overcomes the limitations of previous methods, offering a reliable way to capture and study full-length RNA transcripts. This new technique has the potential to enhance our understanding of the transcriptome, paving the way for new discoveries in genomics and beyond.
Carbonell-Sala S, Perteghella T, Lagarde J, Nishiyori H, Palumbo E, Arnan C, Takahashi H, Carninci P, Uszczynska-Ratajczak B, Guigó R. (2024) CapTrap-seq: a platform-agnostic and quantitative approach for high-fidelity full-length RNA sequencing. Nat Commun 15(1):5278. [article]


