

Understanding the complex world of genes and how they are expressed in different organisms has always been a key challenge in biology. The Long-read RNA-Seq Genome Annotation Assessment Project Consortium, a collective of researchers, has taken significant steps to improve our understanding by focusing on the transcriptome, which is the complete set of RNA transcripts produced by the genome.
What is Long-Read RNA Sequencing?
RNA sequencing (RNA-Seq) is a powerful technique used to study gene expression. Traditional methods, known as short-read sequencing, provide lots of small pieces of data that need to be pieced together like a jigsaw puzzle. Long-read RNA sequencing, on the other hand, offers much longer pieces of data, giving a more comprehensive and detailed view of the RNA transcripts. Think of it like comparing a high-definition panoramic photo to a series of close-up snapshots.
The Consortium’s Mission
The Consortium aimed to evaluate how effective these long-read sequencing methods are in analyzing the transcriptome. They did this by generating an immense amount of data—over 427 million long-read sequences—from different species including humans, mice, and manatees. This data came from various protocols and sequencing platforms to ensure a comprehensive analysis.
Overview of the LRGASP
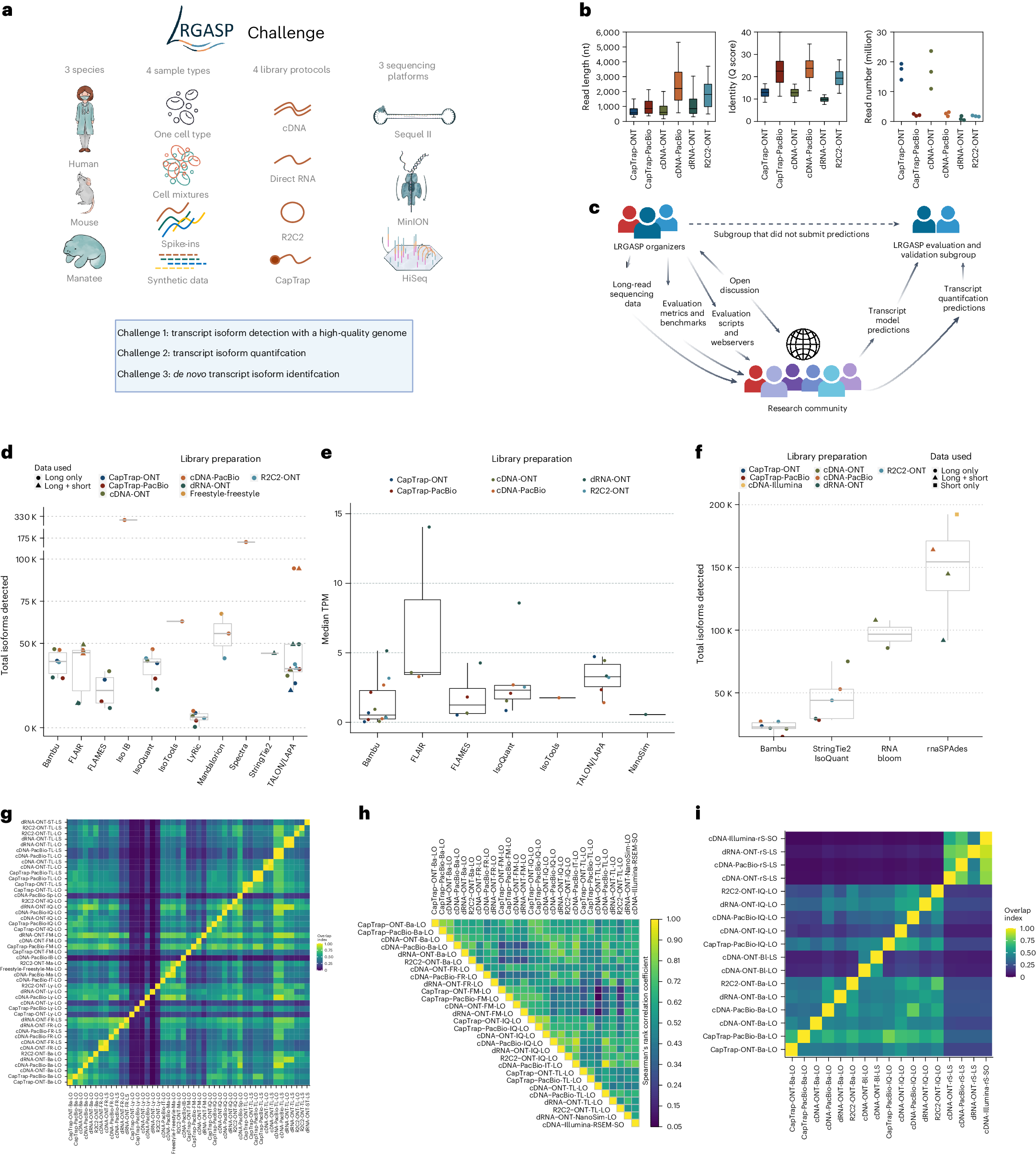
a, Data produced for LRGASP. b, Distribution of read lengths, identify Q score and sequencing depth (per biological replicate) for the WTC11 sample. c, The collaborative design of the LRGASP organizers and participants. d, Number of isoforms reported by each tool on different data types for the human WTC11 sample for Challenge 1. Number of submissions per tool, in order, n = 6, 6, 4, 1, 6, 1, 6, 3, 1, 1 and 12. e, Median TPM value reported by each tool on different data types for the human WTC11 sample for Challenge 2. Number of submissions per tool, in order, n = 11, 3, 4, 6, 1, 6 and 1. f, Number of isoforms reported by each tool on different data types for the mouse ES data for Challenge 3. Number of submissions per tool, in order, n = 6, 5, 2 and 4. g, Pairwise relative overlap of unique junction chains (UJCs) reported by each submission. The UJCs reported by a submission are used as a reference set for each row. The fraction of overlap of UJCs from the column submission is shown as a heatmap. For example, a submission that has a small subset of many other UJCs from other submissions will have a high fraction shown in the rows but a low fraction by column for that submission. Data are only shown for WTC11 submissions. h, Spearman correlation of TPM values between submissions to Challenge 2. i, Pairwise relative overlap of UJCs reported by each submission. The UJCs reported by a submission are used as a reference set for each row. The fraction of overlap of UJCs from the column submission is shown as a heatmap. Ba, Bambu; Bl, RNA-Bloom; FM, FLAMES; FR, FLAIR; IB, Iso_IB; IQ, IsoQuant; IT, IsoTools; Ly, LyRic; Ma, Mandalorion; rS, rnaSPAdes; Sp, Spectra; ST, StringTie2; TL, TALON-LAPA.
Key Findings
- Accuracy Over Depth: The study found that libraries containing longer and more accurate sequences resulted in more precise identification of transcripts compared to those with just a higher number of reads. In simpler terms, quality trumps quantity when it comes to getting accurate transcript information.
- Quantification and Depth: While accuracy is crucial for identifying transcripts, having more reads (greater depth) improves the accuracy of measuring how much of each transcript is present. This means that to understand the abundance of different RNA molecules, having lots of data points helps.
- Reference-Based Tools Shine: In genomes that are already well-documented, tools that rely on these reference sequences performed the best. These tools use existing knowledge about the genome to improve their analysis, making them very effective for well-studied organisms.
- Discovering the Rare and Novel: For finding rare and previously unknown transcripts, the study suggests using additional types of data and replicate samples. This means combining different data sources and repeating experiments to ensure that rare findings are accurate and reliable.
Implications for Future Research
This study serves as a benchmark for current methods in transcriptome analysis. It not only highlights the strengths and limitations of long-read RNA sequencing but also provides valuable insights for future developments in this field. By understanding these findings, researchers can refine their techniques to better explore the complexities of gene expression.
Conclusion
The work of the Long-read RNA-Seq Genome Annotation Assessment Project Consortium represents a significant advancement in the field of transcriptomics. By demonstrating the importance of sequence accuracy, read depth, and the use of reference-based tools, this study guides researchers toward more effective and precise methods for studying RNA transcripts. As we continue to explore the depths of the transcriptome, these insights will be crucial for uncovering the intricate details of gene expression and its impact on biology.
Pardo-Palacios FJ, Wang D, Reese F, Diekhans M, Carbonell-Sala S, Williams B, Loveland JE, De María M, Adams MS, Balderrama-Gutierrez G, Behera AK, Gonzalez Martinez JM, Hunt T, Lagarde J, Liang CE, Li H, Meade MJ, Moraga Amador DA, Prjibelski AD, Birol I, Bostan H, Brooks AM, Çelik MH, Chen Y, Du MRM, Felton C, Göke J, Hafezqorani S, Herwig R, Kawaji H, Lee J, Li JL, Lienhard M, Mikheenko A, Mulligan D, Nip KM, Pertea M, Ritchie ME, Sim AD, Tang AD, Wan YK, Wang C, Wong BY, Yang C, Barnes I, Berry AE, Capella-Gutierrez S, Cousineau A, Dhillon N, Fernandez-Gonzalez JM, Ferrández-Peral L, Garcia-Reyero N, Götz S, Hernández-Ferrer C, Kondratova L, Liu T, Martinez-Martin A, Menor C, Mestre-Tomás J, Mudge JM, Panayotova NG, Paniagua A, Repchevsky D, Ren X, Rouchka E, Saint-John B, Sapena E, Sheynkman L, Smith ML, Suner MM, Takahashi H, Youngworth IA, Carninci P, Denslow ND, Guigó R, Hunter ME, Maehr R, Shen Y, Tilgner HU, Wold BJ, Vollmers C, Frankish A, Au KF, Sheynkman GM, Mortazavi A, Conesa A, Brooks AN. (2024) Systematic assessment of long-read RNA-seq methods for transcript identification and quantification. Nat Methods [Epub ahead of print]. [article]


