

Where the complexity of biological data meets the challenges of noise and high dimensionality, researchers are constantly seeking more effective tools to extract meaningful insights. A recent breakthrough by researchers at Institute for Basic Science, Korea comes in the form of scLENS, a novel dimensionality reduction tool designed to overcome longstanding obstacles and empower researchers with greater accuracy and efficiency in single-cell RNA sequencing (scRNA-seq) data analysis.
The field of scRNA-seq holds immense promise for unraveling the intricacies of cellular behavior and uncovering novel biological phenomena. However, the sheer volume of data and inherent noise present significant hurdles to unlocking its full potential. Traditional dimensionality reduction methods, while valuable, often require manual intervention and are susceptible to signal distortion, leading to biased results.
scLENS is a cutting-edge tool developed by researchers to address these challenges head-on. Unlike its predecessors, scLENS leverages innovative techniques to automatically detect and mitigate signal distortion caused by common data preprocessing methods such as log normalization. By applying L2 normalization to uniformize cell vector lengths, scLENS ensures more accurate representation of biological signals without user bias.
Overview of scLENS
(single-cell Low-dimensional embedding using the effective Noise Subtract)
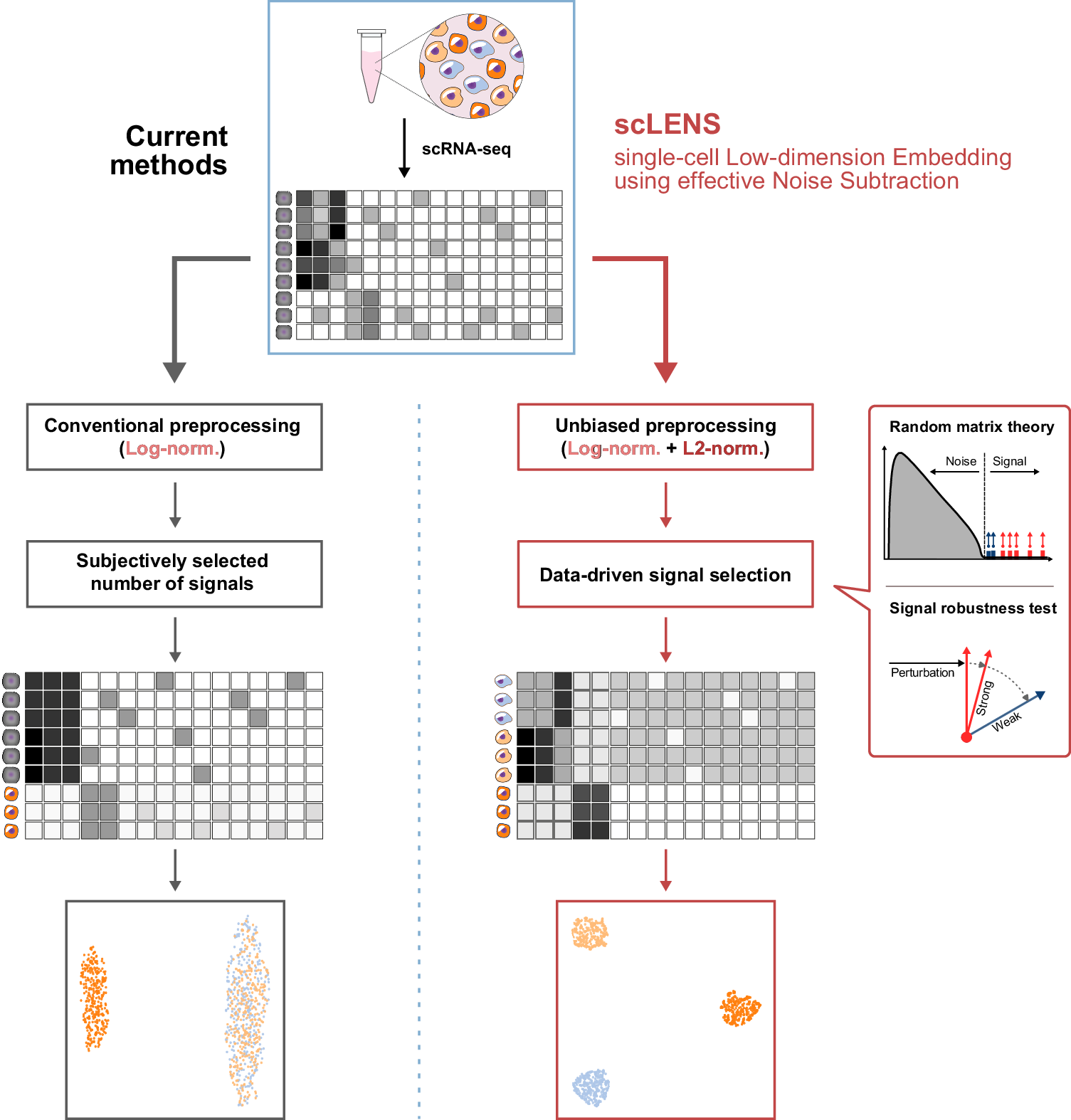
Current dimensionality reduction methods employ log normalization for data preprocessing, which can distort signals in data due to the high level of sparsity and variance between cells (left). They then reduce the data using various dimensionality reduction algorithms. However, during this process, the majority of current methods require the user’s decision to set a threshold to differentiate signals from noise. Due to signal distortion and manual signal selection, current methods often fail to capture the high-dimensional data structure. In contrast, scLENS can prevent signal distortion by incorporating L2 normalization (right). Furthermore, scLENS uses random matrix theory-based noise filtering and signal robustness test-based filtering to automatically select signals without manual selection. As a result, scLENS can perform accurate dimensionality reduction without user bias.
But scLENS doesn’t stop there. Building upon random matrix theory-based noise filtering and signal robustness testing, scLENS offers a data-driven approach to determine the optimal threshold for signal dimensions. This means that researchers can now rely on objective criteria to identify biologically relevant signals, eliminating the need for manual tuning and saving valuable time in the analysis process.
In rigorous testing against 11 widely used dimensionality reduction tools, scLENS has demonstrated superior performance, particularly in challenging scRNA-seq datasets characterized by high sparsity and variability. Its ability to accurately detect signals and reduce noise makes scLENS a powerful ally for researchers seeking deeper insights into cellular dynamics and biological processes.
To further streamline its accessibility and usability, the developers of scLENS have made it available as a user-friendly package, equipped with automated features for precise signal detection in scRNA-seq data. This democratization of advanced analytical tools promises to accelerate discoveries in the field of single-cell genomics and pave the way for transformative breakthroughs in our understanding of cellular biology.
Availability – The Julia codes for the scLENS, and the codes for the R and Python packages used in the benchmark study are available in the Mathbio GitHub: https://github.com/Mathbiomed/scLENS.
Kim H, Chang W, Chae SJ, Park JE, Seo M, Kim JK. (2024) scLENS: data-driven signal detection for unbiased scRNA-seq data analysis. Nat Commun 15(1):3575. [article]


