

Cells often alter metabolic strategies under nutrient-deprived conditions to support their survival and growth. Characterizing metabolic reprogramming in the tumor microenvironment (TME) is of emerging importance in cancer research and patient care. However, recent technologies only measure a subset of metabolites and cannot provide in situ measurements. Computational methods such as flux balance analysis (FBA) have been developed to estimate metabolic flux from bulk RNA-seq data and can potentially be extended to single-cell RNA-seq (scRNA-seq) data. However, it is unclear how reliable current methods are, particularly in TME characterization.
Researchers at the University of Texas MD Anderson Cancer Center have developed a computational framework METAFlux (METAbolic Flux balance analysis) to infer metabolic fluxes from bulk or single-cell transcriptomic data. Large-scale experiments using cell-lines, the cancer genome atlas (TCGA), and scRNA-seq data obtained from diverse cancer and immunotherapeutic contexts, including CAR-NK cell therapy, have validated METAFlux’s capability to characterize metabolic heterogeneity and metabolic interaction amongst cell types.
The workflow of METAFlux
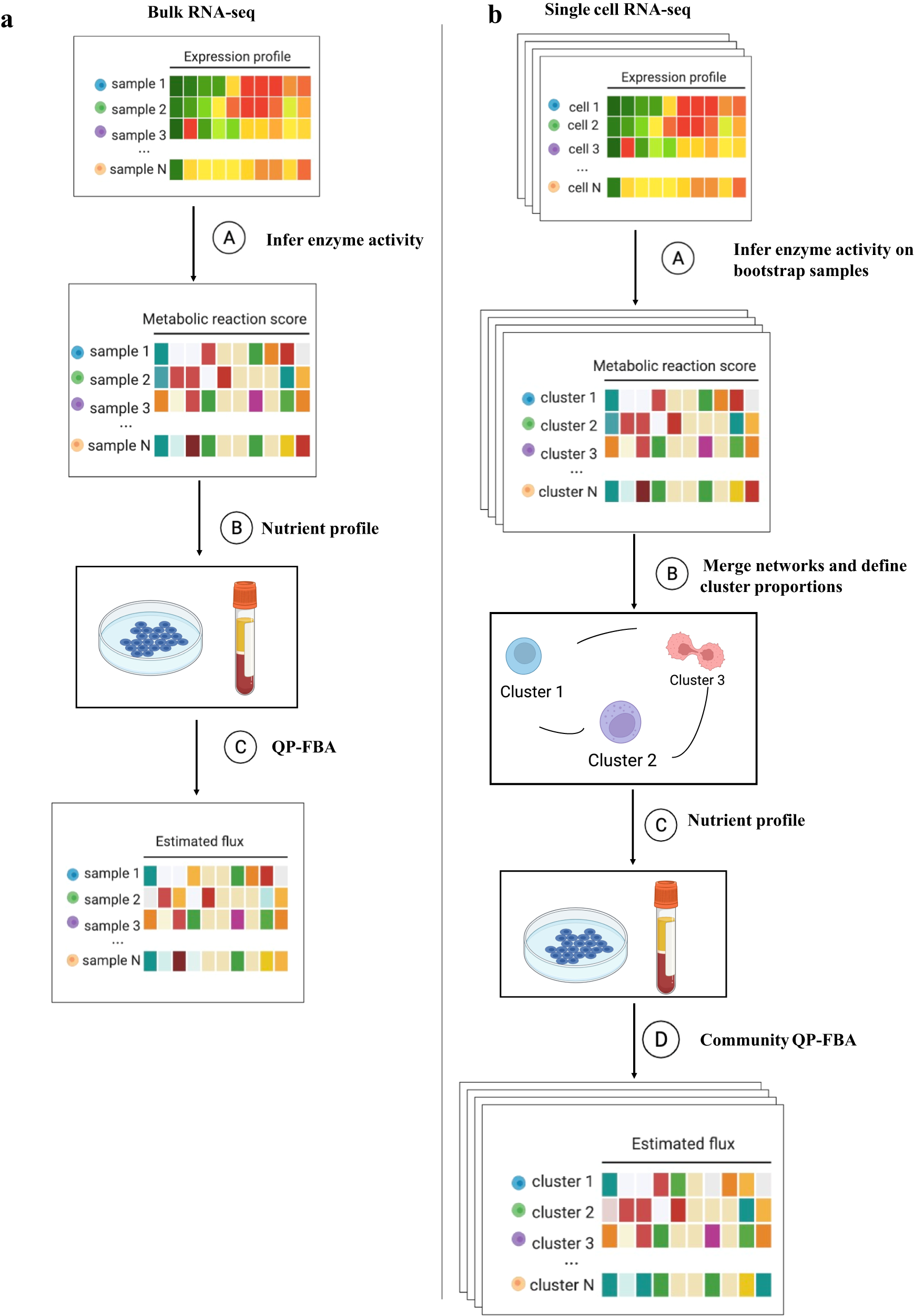
a The workflow of METAFlux in bulk RNA-seq setting. In step A, metabolic reaction activity scores (MRAS) are estimated from RNA-seq data. In step B, a nutrient profile is defined so only certain nutrients can be uptaken. In step C, quadratic programming-based FBA (flux balance analysis) is performed to estimate metabolic fluxes for each sample. The figure was created with BioRender.com. b The workflow of METAFlux in single-cell RNA-seq setting. In step A, metabolic reaction activity scores (MRAS) are estimated for each stratified bootstrap sampled single-cell dataset. In step B, metabolic networks for different clusters are merged to form one community, and proportions of clusters should be defined during this step. In step C, nutrient profile is defined so only specific metabolites can be uptaken by TME. In step D, community-based quadratic programming FBA is constructed to estimate per cell average metabolic fluxes for each cluster and total average metabolic fluxes for overall TME.
Availability – Source code is publicly available at https://github.com/KChen-lab/METAFlux or https://zenodo.org/badge/latestdoi/515741372.
Huang Y, Mohanty V, Dede M, Tsai K, Daher M, Li L, Rezvani K, Chen K. (2023) Characterizing cancer metabolism from bulk and single-cell RNA-seq data using METAFlux. Nat Commun 14(1):4883. [article]


