
RNA-seq followed by de novo transcriptome assembly has been a transformative technique in biological research of non-model organisms, but the computational processing of RNA-seq data entails many different software tools. The complexity of these de novo transcriptomics workflows therefore presents a major barrier for researchers to adopt best-practice methods and up-to-date versions of software.
A team led by researchers at UC San Diego has developed a streamlined and universal de novo transcriptome assembly and annotation pipeline, transXpress, implemented in Snakemake. transXpress supports two popular assembly programs, Trinity and rnaSPAdes, and allows parallel execution on heterogeneous cluster computing hardware.
A schema of the data processing steps performed by the transXpress pipeline
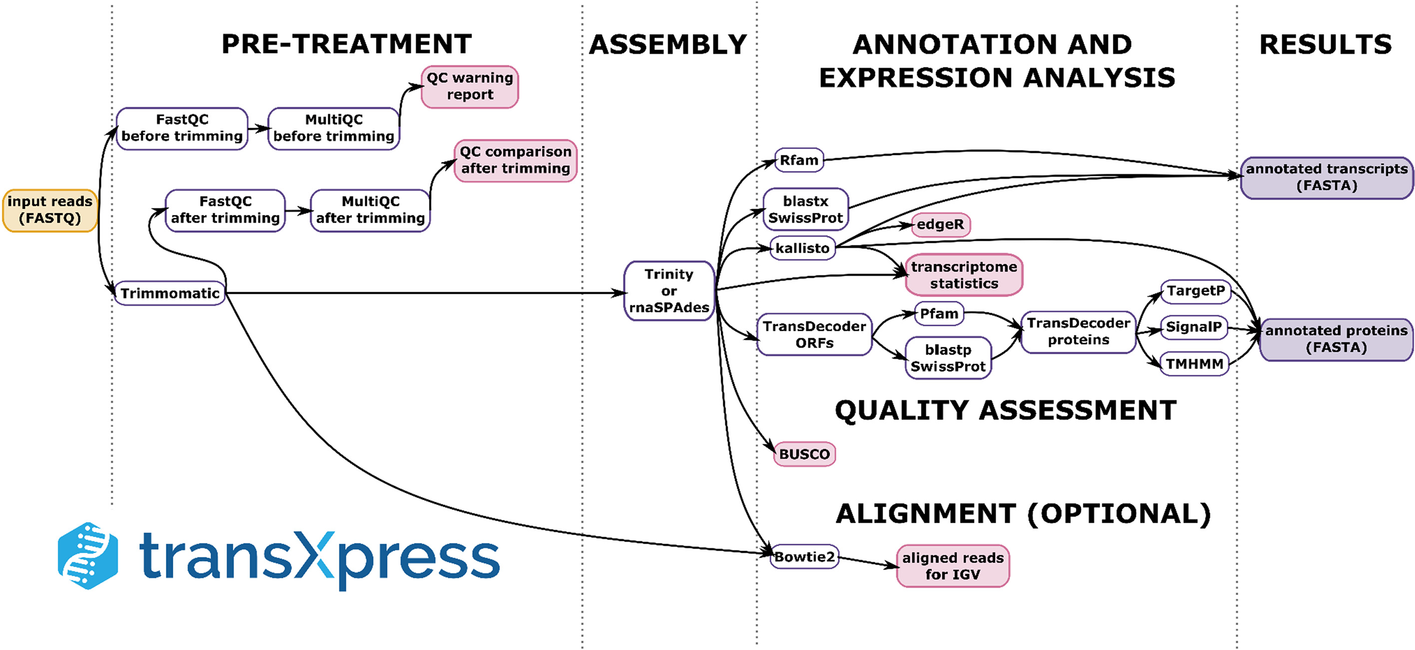
The input data are on the very left in a yellow-colored frame. Initial data pre-treatment tasks are on the left, followed by assembly and tasks executed largely in parallel (annotation and expression analysis). Output data types are in a purple background on the very right. This is a manually simplified version of the directed acyclic graph (DAG) of Snakemake tasks. The DAG can be automatically generated by Snakemake for each transXpress run
transXpress simplifies the use of best-practice methods and up-to-date software for de novo transcriptome assembly, and produces standardized output files that can be mined using SequenceServer to facilitate rapid discovery of new genes and proteins in non-model organisms.
Availability – Project home page: https://github.com/transXpress/transXpress
Fallon TR, Čalounová T, Mokrejš M, Weng JK, Pluskal T. (2023) transXpress: a Snakemake pipeline for streamlined de novo transcriptome assembly and annotation. BMC Bioinformatics 24(1):133. [article]


