

In a study published in Nature Communications, Prof. CHAO Yanjie’s group at the Institute of Shanghai Institute of Immunity and Infection of the Chinese Academy of Sciences reported a novel RNA interactome profiling technology (iRIL-seq), which identifies RNA-RNA molecular interactions in live microbial cells. Using iRIL-seq, the researchers for the first time mapped the RNA-RNA interactome networks of Salmonella enterica at different growth stages, and discovered many novel non-coding RNAs and important mRNA regulatory hubs.
Direct RNA-RNA interaction is a main mechanism by which non-coding RNAs exert their functions. In prokaryotic cells, non-coding small RNAs recognize target mRNAs through very short and imperfect basepairing interactions, which are difficult to be predicted and identified. Bacterial non-coding RNAs have important regulatory functions, controlling the expression of many target genes at the post-transcriptional level, and playing crucial roles in many biological processes, such as bacterial infection, antibiotic resistance, central metabolism, stress response, quorum sensing, and biofilm formation.
The analysis of the RNA-RNA interaction network is of great significance to the study of microbial physiology and the functions of non-coding RNAs. However, the existing RNA-RNA interactome profiling techniques are complicated, which require fixed cells and numerous in vitro steps such as RNA digestion, repair and ligation, yielding insufficient RNA interactions with low resolution and accuracy.
In this study, the researchers developed a novel in vivo RNA proximity-ligation approach, iRIL-seq (intracellular RNA interaction by ligation and sequencing).
The researchers first induced the expression of T4 RNA ligase in living microbial cells, which leads to the proximity-ligation between interacting RNA molecules in vivo. They then enriched the non-coding small RNAs and their interacting transcripts by immunoprecipitation of the major small RNA chaperone Hfq. Finally they determined the identity and sequence of the interacting RNA molecules using high-throughput RNA sequencing. The main experimental process can be completed within a single day, thanks to the rapid ligation in vivo and streamlined workflow.
This new technology not only illustrates the atlas of microbial RNA-RNA interaction network, but also identifies RNA-RNA basepairing interactions at the single-nucleotide resolution. The iRIL-seq technology is a simple, fast, accurate and universal technology for analyzing the RNA-RNA interactions in microorganisms, and proposes a new way to study RNA-RNA interactions in eukaryotic cells.
iRIL-seq faithfully captures sRNA-target interactions
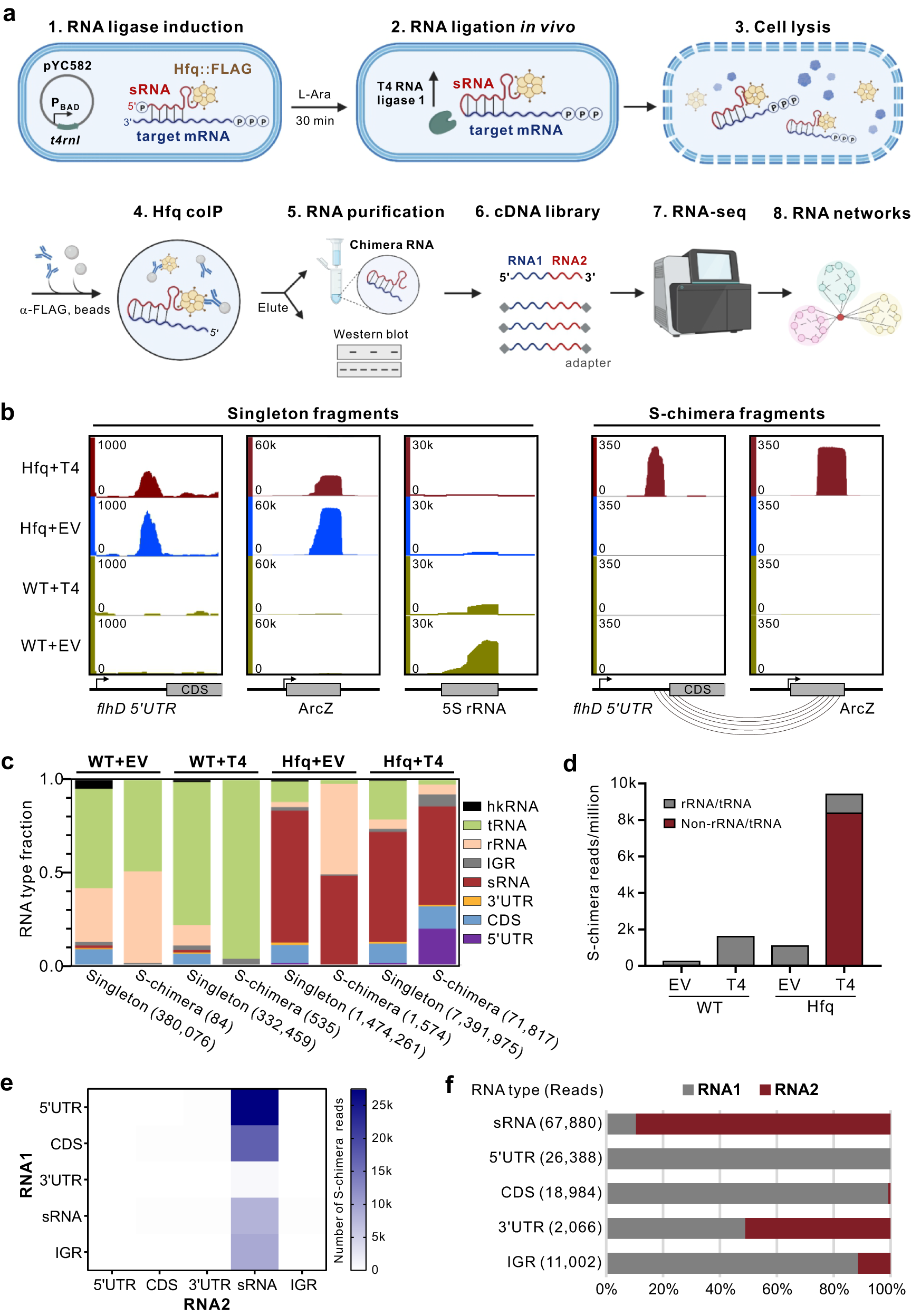
a Schematic of iRIL-seq. Salmonella Hfq::FLAG strain carrying the plasmid pBAD-t4rnl1 (pYC582) was grown in LB. sRNA-target pairs were ligated to form chimeras in vivo by T4 RNA ligase induced for 30 min with L-arabinose. The ligation chimeras bound to 3xFLAG tagged Hfq were enriched using coIP from bacterial lysates. Chimeras were then purified, identified by deep sequencing, and used to determine the RNA interaction network by subsequent in silico analysis. Created with BioRender.com. b iRIL-seq captured known sRNA-target interactions. Genome browser screenshots showing the genomic locations of indicated RNAs covered by singleton or significant chimera reads (S-chimera, p < 0.05, one-sided Fisher’s exact test). Salmonella WT and Hfq::FLAG strains carrying empty vector or pYC582 were grown in LB. Bacteria were treated with L-arabinose for 30 min and grown to OD600 of 2.0. Cells were collected and performed iRIL-seq. EV: empty vector. T4: pBAD-t4rnl1 (pYC582). ORFs and RNAs were indicated by gray boxes. c Distribution of each transcript type for singleton and S-chimeric fragments within one set of four iRIL-seq libraries. The total number of sequenced fragments was denoted in parentheses. hkRNA: four housekeeping RNA (RnpB, SsrS, Ffs and SsrA). IGR: intergenic region. EV and T4 are the same as in (b). d Number of S-chimeric fragments detected in each sample. EV and T4 are the same as in (b). e Number of S-chimeric fragments for different transcript types. RNA1, the 5’ terminal RNA in the chimera. RNA2, the 3’ terminal RNA in the chimera. f Distribution of RNA1 and RNA2 in S-chimeras for different transcript types.
Furthermore, the researchers mapped for the first time the dynamic RNA-RNA interactomes of bacterial pathogen Salmonella enterica at multiple growth stages, identified more than 2,000 RNA-RNA interactions, and discovered many key mRNA regulatory hubs. The largest mRNA regulatory hub in bacteria to date was shown to be the mRNA encoding the outer membrane porin OmpD, which is regulated by at least 12 different non-coding small RNAs.
Experimental characterization of these small RNAs led to the identification of a novel small RNA FadZ that is cleaved from an mRNA 3’UTR by ribonuclease. The expression of FadZ and its parental fadBA mRNA were regulated by the upstream transcription factors FadR, CRP, and long-chain fatty acid signals. Under long-chain fatty acid conditions, both FadZ sRNA and its target gene ompD were activated by the transcription factor CRP, together constituting a type I incoherent feed-forward loop (I1-FFL). The finding revealed the post-transcriptional regulatory mechanism of bacteria in response to long-chain fatty acid metabolism in hosts.
Source – Chinese Academy of Sciences
Liu F, Chen Z, Zhang S, Wu K, Bei C, Wang C, Chao Y. (2023) In vivo RNA interactome profiling reveals 3’UTR-processed small RNA targeting a central regulatory hub. Nat Commun 4(1):8106. [article]


