

It is said in Spanish, “si no existe, créalo” or “if it does not exist, create it.” This is exactly what sometimes occurs at the Fred Hutch. When we do not have the tools to test a hypothesis, we must be creative! A great example is a collaboration between Dr. Susan Bullman, an Assistant professor of Human Biology, and Dr. Christopher Johnson, an assistant professor of Vaccine and Infectious Disease, that resulted in a recent Nature Protocols publication. The research teams developed a method called INVADEseq (invasion-adhesion-directed expression sequencing) for identifying bacterial transcripts in cancer cells.
A growing number of human cancer types are characterized by the presence and composition of a tumor microbiome, a collection of cancer-associated bacterial species that add yet another layer of complexity to this disease and can influence prognosis or treatment response. Until recently, researchers studying tumor samples have primarily relied on next-generation sequencing-based methods, such as single-cell RNA sequencing (scRNAseq) to study the host cells’ transcriptional profiles. However, scRNAseq generally fails to detect bacteria within tumor samples, since this method relies on the poly(A) tail at the 3′ end of the transcripts that are present only in eukaryotic mRNAs. As bacterial RNAs generally lack the poly(A) tail, scRNAseq cannot identify bacteria within or those associated with a cancer cell.
So, how can we identify bacterial transcripts within a tumor sample? To answer this question, Dr. Jorge Galeano Niño, a postdoctoral research fellow in the Bullman lab and leading author of the study, took advantage of the fact that ~90% of the bacterial transcriptome is either 16S or 23S ribosomal RNA (rRNA). Since the 16S gene has universally conserved regions across microbial communities, the team developed a new method, the INVADEseq protocol, which introduces a primer that targets the bacterial 16S rRNA gene.
INVADEseq was developed using the 10X Genomics Chrominum 5′ scRNA assay, which utilizes the SMART technology (Switching Mechanism at the 5′ end of RNA Template) to construct the cDNA libraries. This technology relies on the template switching activity of Moloney murine leukemia virus reverse transcriptase (MMLV RT) to synthesize and anchor the first-strand cDNA to the gel beads-in-emulsions (GEMs). By using the MMLV RT, the mRNA is first transcribed into cDNA using a primer that binds to the eukaryotic poly(A) mRNA. Additionally, this MMLV RT adds three non-template nucleotides to the 5′ end of the newly synthesized first-strand cDNA, which binds to complementary nucleotides from a template switching oligo (TSO) that also contains a 10x barcode sequence and a unique molecular identifier (UMI) that can be used to scale down for the amplification steps thus providing the original transcriptional load. Using this method, the template strands can be switched from cellular RNA to TSO, which serve as primer-binding sites for the subsequent amplification steps. Since bacterial RNA transcripts usually lack poly(A) tails, this approach is not sufficient to detect bacterial transcripts. Here is the trick! The team used the same SMART technology that was implemented to capture poly(A) transcripts to capture bacterial transcripts contained in the GEMs by priming the conserved regions of the bacterial 16S rRNA gene. The subsequent reverse transcription of the variable regions of the 16S rRNA gene allowed the team to taxonomically resolved the bacterial communities that were associated with a single eukaryotic cell.
Tumor processing for single-cell RNAseq acquisition and computational pipeline for host and bacteria cell annotations, host-associated transcriptome and GSEA pathway enrichment analysis
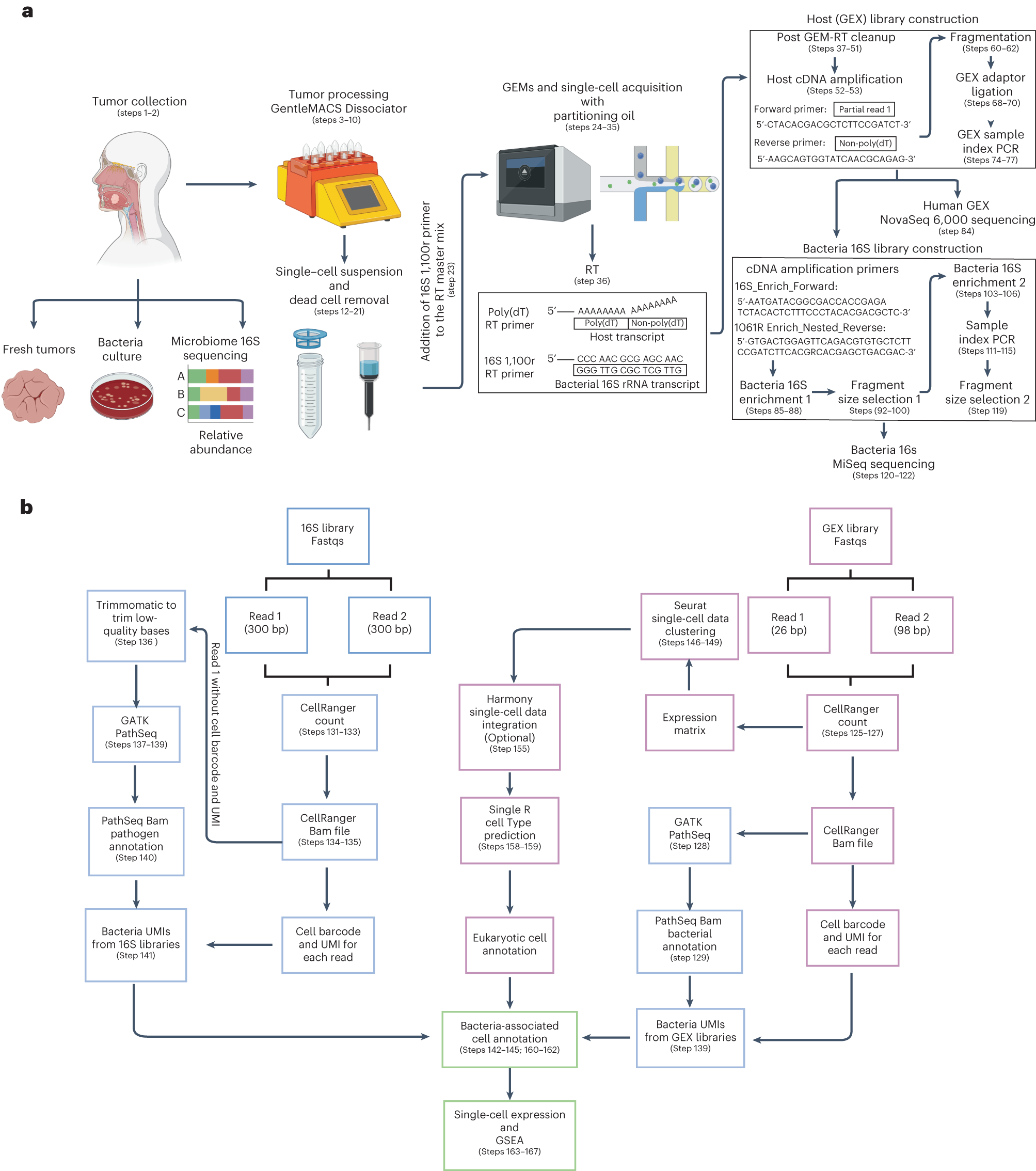
a, Tumor samples were isolated from patients with gastrointestinal tract cancers. Bacteria culture in blood agars and microbiome 16S rRNA sequencing analysis were performed to screen tumor samples that were positive for bacteria. To obtain single-cell suspensions, tumor samples were processed using the gentleMACS-quality Octo Dissociator equipped with electrical heaters. The cell suspension was passed through a 70-µm cell strainer and dead cells were removed by magnetic sorting using LS columns. Single-cell suspensions were loaded onto a Chromium Chip K and processed with the 10x Chromium controller to capture single cells within a gel bead emulsion (GEM) containing a master mix with two primers, one that targets the polyadenylated host mRNA and second that targets the bacterial 16S rRNA gene. Following RT, the hosts (GEX) cDNA libraries were prepared and sequenced using the NovaSeq 600 platform. An aliquot from the GEX cDNA libraries were acquired to enriched for bacteria transcripts by amplifying the bacterial 16S rRNA gene. Using the BluePippin system fragment sizes between 955 and 1,215 bp were selected generating the bacteria 16S libraries that were sequenced using the MiSeq platform. b, Reads from the GEX libraries were mapped with the human reference genome GRCh38 using Cellranger Count. Then, the unmapped GEX reads with an adequate cell barcode and UMI count were processed via GATK PathSeq, thus obtaining bacterial UMI matrices for each bacteria-associated single cell. Reads from the 16S bacterial enrichment libraries were processed using Cellranger Count to obtain the corresponding barcode and UMI. Then R1 reads without a barcode or UMI were trimmed to remove low-quality bases and converted to BAM files to process through GATK PathSeq obtaining the bacteria UMI matrix for valid host cells from the GEX libraries. The bacterial UMI matrices from the GEX and 16S bacterial enrichment libraries were merged, removing UMI duplicates. Single-cell expression matrices from the GEX libraries were processed by Seurat followed by SingleR package software to obtain the annotations for each eukaryotic cell cluster. Harmony software was used to integrate single-cell datasets when it was required. The merged bacteria matrix was attached to the single-cell data identifying the host single cells that harbored bacterial transcripts. Gene expression profile and GSEA pathway enrichment analyses were performed based on the presence or absence of bacteria, at various taxonomic levels, at host single-cell-level resolution.
To test their technology, the team identified cell-associated bacteria in oral squamous cell carcinoma (OSCC) tumor tissue, which is thought to be influenced by the microbiome. By using INVADEseq, the researchers demonstrated that cell-associated bacteria were largely present inside a subset of epithelial and macrophage single cells. Moreover, the team found that Fusobacterium and Treponema species were the predominant bacteria associated with OSCC tumors. The INVADEseq approach has been shown to be an effective method for identifying and analyzing bacterial transcriptomics from tissue samples at a single cell level. More broadly, this tool will enable researchers to investigate the host gene signatures associated to certain bacteria species at the single cell level to further our understanding of how these relationships influence cancer initiation and progression.
Source – Fred Hutch
Galeano Niño JL, Wu H, LaCourse KD, Srinivasan H, Fitzgibbon M, Minot SS, Sather C, Johnston CD, Bullman S. (2023) INVADEseq to identify cell-adherent or invasive bacteria and the associated host transcriptome at single-cell-level resolution. Nat Protoc 18(11):3355-3389. [article]


