

Understanding how individual cells operate and interact is crucial. One powerful technique that scientists use to explore this cellular universe is single-cell transcriptome sequencing, or scRNA-seq. This high-tech method allows researchers to study gene expression in individual cells, providing a detailed map of cellular activity. However, analyzing scRNA-seq data comes with its own set of challenges, particularly when it comes to accurately grouping cells into different types based on their gene expression profiles.
The Challenge of Clustering in scRNA-seq Data
When scientists analyze scRNA-seq data, one of the first steps is clustering. This process involves grouping cells that have similar gene expression patterns, which helps identify different cell types and understand their interactions. Think of it as organizing a library where each book represents a cell, and you want to group books by their genres (cell types). However, unlike neatly organized libraries, scRNA-seq data is messy. There’s a lot of noise (irrelevant or misleading data), and the complex nature of gene expression makes it difficult to find the right “genres” for each “book.”
Introducing scRISE: A New Approach to Clustering
To address these challenges, researchers at the East China University of Science and Technology have developed a new deep clustering method called scRISE (scRNA-seq Iterative Smoothing and self-supervised discriminative Embedding model). This innovative approach combines two powerful techniques to improve the quality of data representation and clustering:
- Iterative Smoothing Module: This module uses graph autoencoders, which are advanced algorithms designed to handle complex data structures. The goal here is to reduce the noise in the data and refine the pairwise similarity between cells. By iteratively smoothing the data, scRISE gradually incorporates the structural features of cells, enriching the information available for clustering.
- Self-supervised Discriminative Embedding Module: This module works to partition cells into the correct clusters by using an adaptive similarity threshold. It’s like having a smart librarian who can adjust the criteria for sorting books based on the evolving structure of the library. This self-supervised approach ensures that cells are grouped more accurately, even as new data is added.
The overview of the proposed method scRISE
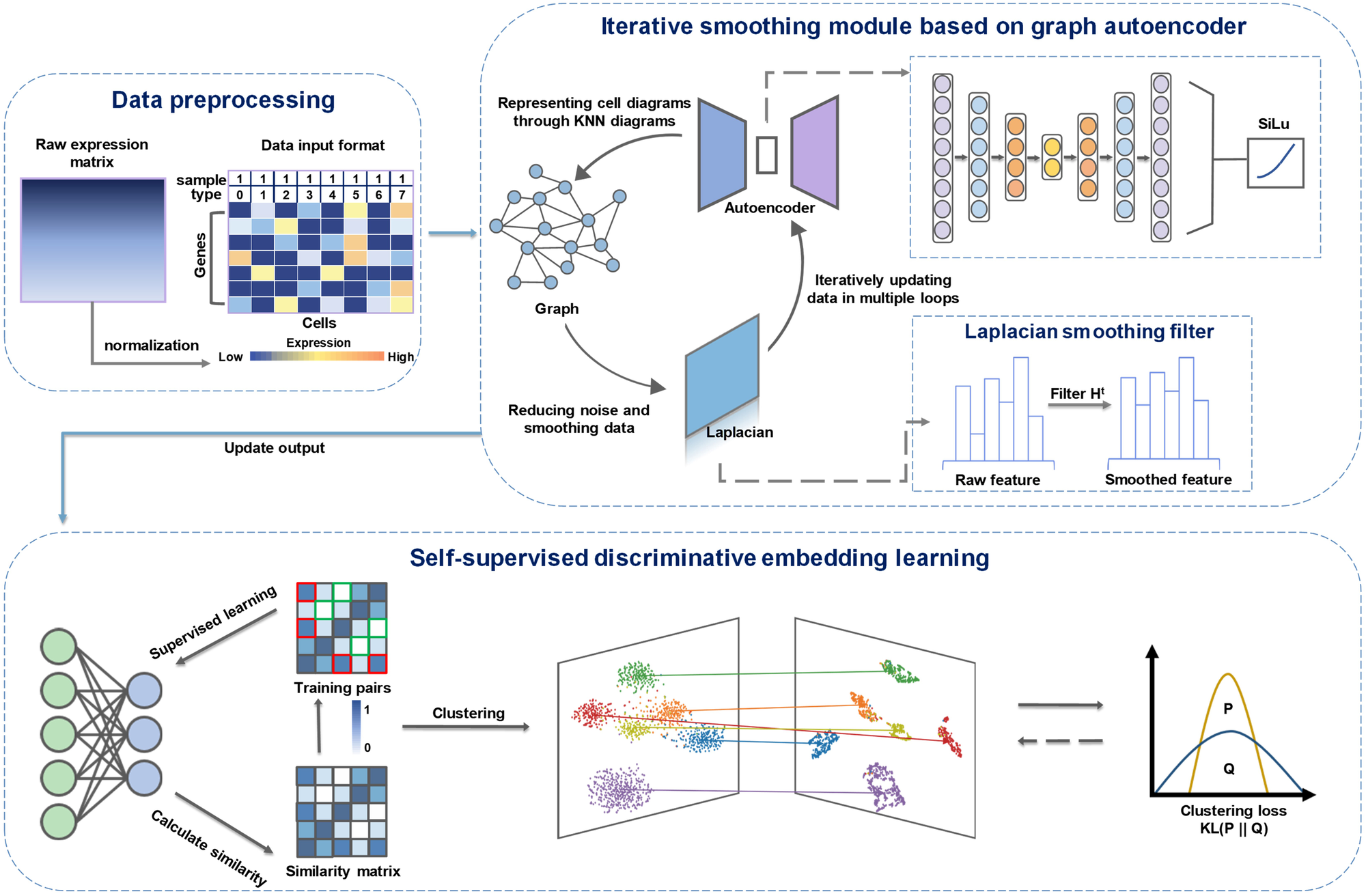
The framework of scRISE includes the iterative smoothing module based on graph autoencoder and the self-supervised discriminative embedding learning module. The iterative smoothing module consists of an autoencoder and a Laplacian filter connected. In each iteration, a cell graph is constructed from the input data, and the reconstructed data from the autoencoder is smoothed using the Laplacian filter. The smoothed data are fed back to the autoencoder for further processing. The output of the autoencoder is then processed through the self-supervised discriminative embedding module, which adopts an adaptive threshold to identify positive and negative sample pairs to compute the final clustering.
The Power of scRISE in Action
The scRISE method has been tested on seventeen different scRNA-seq datasets and has shown significant improvements over other state-of-the-art deep learning clustering methods. One particularly exciting application of scRISE was its use in analyzing data from head and neck squamous cell carcinoma (HNSCC), a type of cancer. Through this analysis, scRISE unveiled 62 informative genes, shedding light on their potential roles as therapeutic targets and biomarkers. This means that scRISE not only helps in accurately grouping cells but also in identifying key genes that could be crucial in developing new treatments and diagnostics for diseases.
Conclusion
The development of scRISE represents a significant advancement in the field of single-cell transcriptome sequencing. By effectively reducing noise and refining the clustering process, scRISE provides researchers with a more accurate and detailed understanding of cellular functions and interactions. This, in turn, can lead to the discovery of new therapeutic targets and biomarkers, paving the way for advancements in personalized medicine and treatments for complex diseases like cancer. As we continue to refine these techniques, the possibilities for uncovering the mysteries of the cellular world are boundless, promising a future where we can better understand and manipulate the very building blocks of life.
Availability – The scRISE software package and source code are available in Github (https://github.com/LiLab-ssruan/scRISE).
Xie J, Ruan S, Tu M, Yuan Z, Hu J, Li H, Li S. (2024) Clustering single-cell RNA sequencing data via iterative smoothing and self-supervised discriminative embedding. Oncogene [Epub ahead of print]. [abstract]


