

Most methods for single-cell transcriptome sequencing amplify the termini of polyadenylated transcripts, capturing only a small fraction of the total cellular transcriptome. This precludes the detection of many long non-coding, short non-coding and non-polyadenylated protein-coding transcripts and hinders alternative splicing analysis.
A team led by researchers at the Hubrecht Institute-KNAW has now developed VASA-seq to detect the total transcriptome in single cells, which is enabled by fragmenting and tailing all RNA molecules subsequent to cell lysis. The method is compatible with both plate-based formats and droplet microfluidics. The researchers applied VASA-seq to more than 30,000 single cells in the developing mouse embryo during gastrulation and early organogenesis. Analyzing the dynamics of the total single-cell transcriptome, they discovered cell type markers, many based on non-coding RNA, and performed in vivo cell cycle analysis via detection of non-polyadenylated histone genes. RNA velocity characterization was improved, accurately retracing blood maturation trajectories. Moreover, our VASA-seq data provide a comprehensive analysis of alternative splicing during mammalian development, which highlighted substantial rearrangements during blood development and heart morphogenesis.
Overview of the VASA-seq workflow and benchmarking against
other state-of-the-art methodologies
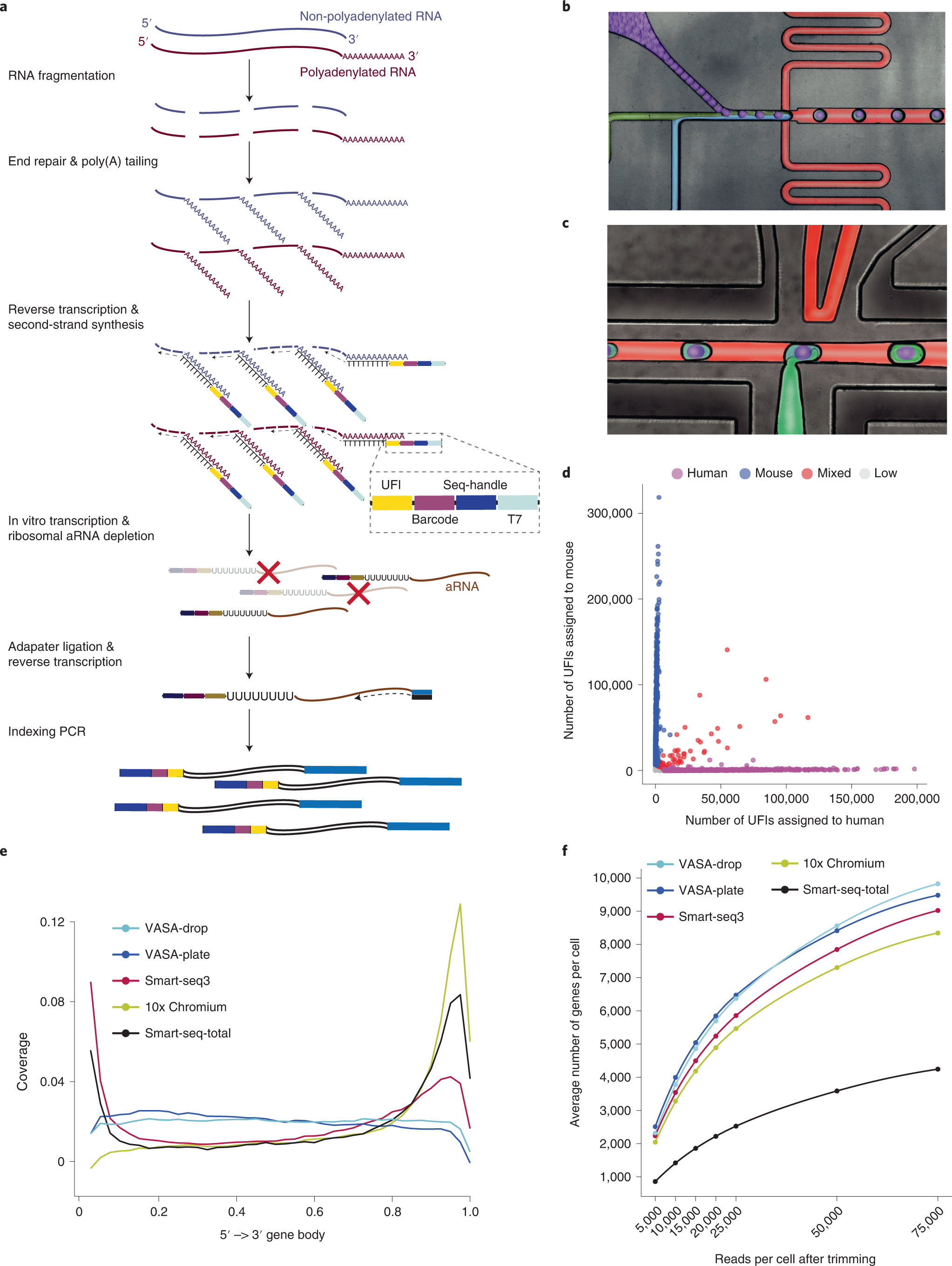
a, Overview of the VASA-seq single-cell molecular workflow. Single cells are lysed, and RNA is fragmented. Fragments are repaired and polyadenylated, followed by reverse transcription (RT) using barcoded oligo-dT primers. The cDNA is made double stranded and amplified using IVT. aRNA is depleted of rRNA, and libraries are finalized by ligation, RT and PCR, which leave fragments ready for sequencing. b, Picture illustrating the single-cell encapsulation process using droplet microfluidics. The single cells (green) are co-encapsulated with a barcoded bead (purple), lysis and fragmentation mix (blue), and compartmentalization is achieved with the addition of fluorinated surfactant oil (red) at the flow-focusing junction. c, Picture illustrating the picoinjection of reagents (green) to single-cell lysates (light blue/purple). The droplet surface tension is perturbed using an electric field that allows for the subsequent additions of end repair/poly(A) and RT mix. d, Cross-contamination test for VASA-drop was carried out using HEK293T cells (human) and mouse embryonic stem cells (mouse). Barcodes with more than 25% of detected UFIs belonging to the other species were considered doublets/mixed (red). Detected barcodes with low UFIs (<7,500) were discarded (gray). The remainder were assigned to either human (magenta) or mouse (blue). e, Gene body coverage comparison along protein-coding genes. VASA-seq showed even coverage, whereas 10x, Smart-seq-total and Smart-seq3 had a bias toward transcript termini (3′ or 5′ and 3′, respectively). f, The number of detected annotated genes in HEK293T cells, for each method, is plotted against the number of reads (after quality filtering, adapter removal and homopolymer trimming) per cell across different downsampling thresholds. The saturation curves showed that VASA-seq was the most sensitive of the methods. Curvature of gene detection indicated that full complexity was not reached for the method when 75,000 reads were allocated to each cell. Only cells that were sequenced to at least 75,000 reads were used (VASA-plate: n = 174, VASA-drop: n = 376, Smart-seq3: n = 113, Smart-seq-total = 260, 10x Chromium: n = 288).
Salmen F, De Jonghe J, Kaminski TS, Alemany A, Parada GE, Verity-Legg J, Yanagida A, Kohler TN, Battich N, van den Brekel F, Ellermann AL, Arias AM, Nichols J, Hemberg M, Hollfelder F, van Oudenaarden A. (2022) High-throughput total RNA sequencing in single cells using VASA-seq. Nat Biotechnol [Epub ahead of print]. [article]


