

Rapid development of transcriptome sequencing technologies has resulted in a data revolution and emergence of new approaches to study transcriptomic regulation such as alternative splicing, alternative polyadenylation, CRISPR knockout screening in addition to the regular gene expression. A full characterization of the transcriptional landscape of different groups of cells or tissues holds enormous potential for both basic science as well as clinical applications. Although many methods have been developed in the realm of differential gene expression analysis, they all geared towards a particular type of sequencing data and failed to perform well when applied in different types of transcriptomic data. To fill this gap, researchers at the University of Nebraska Medical Center have developed a negative beta binomial t-test (NBBt-test). NBBt-test provides multiple functions to perform differential analyses of alternative splicing, polyadenylation, CRISPR knockout screening, and gene expression datasets. Both real and large-scale simulation data show superior performance of NBBt-test with higher efficiency, and lower type I error rate and FDR to identify differential isoforms and differentially expressed genes and differential CRISPR knockout screening genes with different sample sizes when compared against the current very popular statistical methods.
Computational procedures and workflow of NBBt-test
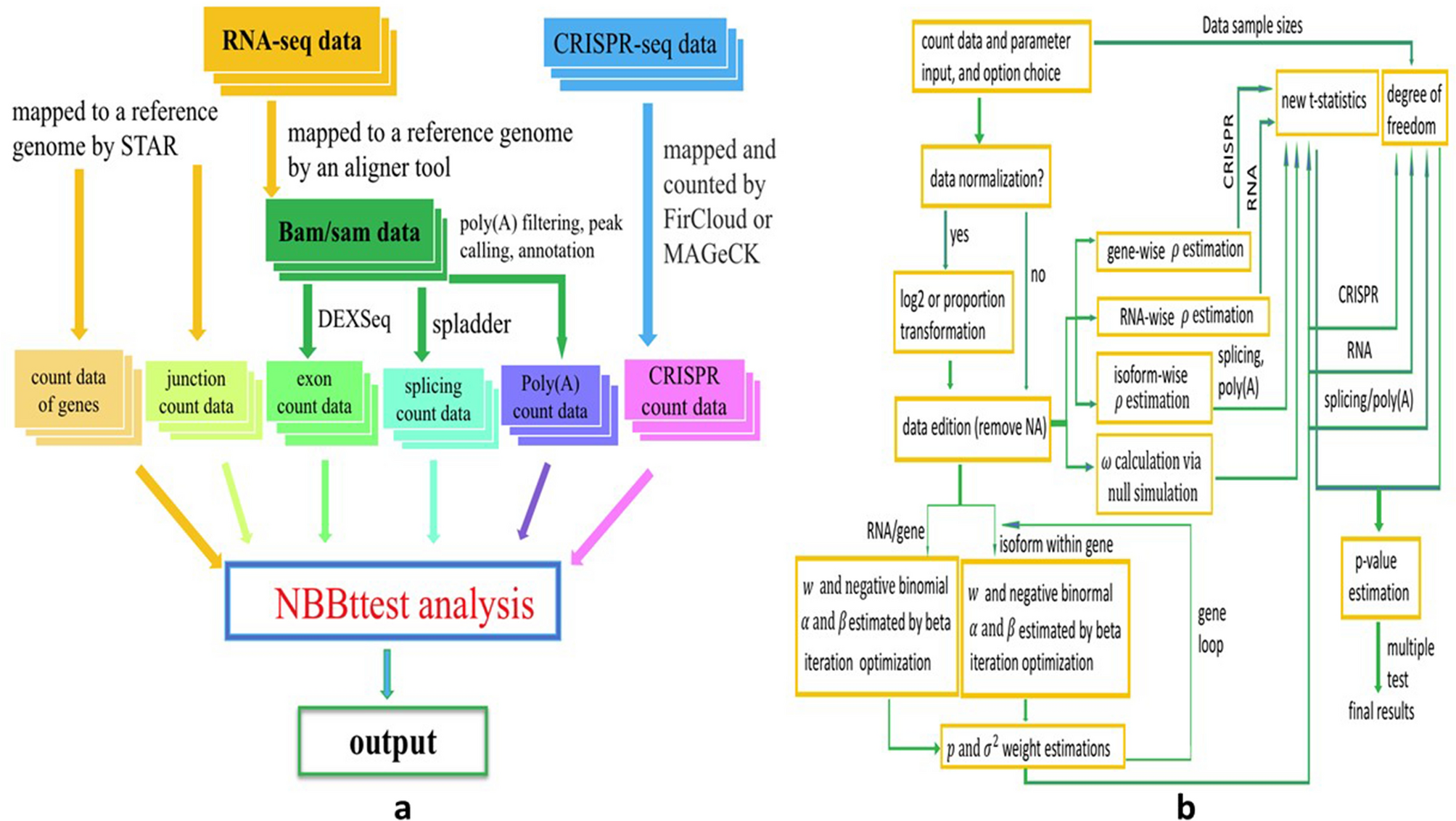
(a) Workflow of NBBt-test. RNA-seq data have two types: RNA-seq reads and CRISPR-seq reads. RNA-seq reads are mapped to a reference genome such as mm9 or mm10 in mice, or hg19(GRCh37) or GRCh38 in human. Currently, popular aligners are STAR, Tophat, BWA, and Boetie. STAR can use RNA-seq data such as fastq and fasta to map RAN-seq reads to reference genome with STAR index. STAR outputs count data of genes, and sorted bam data, For studying splicing, one needs another tool such as spladder, rMATS, cufflinks, to remap bam file to annotated genome file such as hg.gtf or hg.gff3. CRISPR-seq reads are mapped to genomes by MAGeCK to generate count data. (b) Computational procedures. In this workflow, first, the count data (RPKM, FPKM or TPM) is normalized with log2, or proportion transformation (library size is the same across all individuals). Normalized count data are used to estimate ρ and ω and α and β, variance V, and proportion p. Finally, new t-statistics and p-values are calculated to perform multiple tests or adjust p-values.
Availability – An R-package implementing NBBt-test is available for downloading from CRAN (https://CRAN.R-project.org/package=NBBttest).
Tan YD, Guda C. (2022) NBBt-test: a versatile method for differential analysis of multiple types of RNA-seq data. Sci Rep 12(1):12833. [article]


