

Gene expression models, which are key towards understanding cellular regulatory response, underlie observations of single-cell transcriptional dynamics. Although RNA expression data encode information on gene expression models, existing computational frameworks do not perform simultaneous Bayesian inference of gene expression models and parameters from such data. Rather, gene expression models—composed of gene states, their connectivities and associated parameters—are currently deduced by pre-specifying gene state numbers and connectivity before learning associated rate parameters.
Arizona State University researchers propose a method to learn full distributions over gene states, state connectivities and associated rate parameters, simultaneously and self-consistently from single-molecule RNA counts. The researchers propagate noise from fluctuating RNA counts over models by treating models themselves as random variables. They achieve this within a Bayesian non-parametric paradigm. They demonstrate the method on the Escherichia coli lacZ pathway and the Saccharomyces cerevisiae STL1 pathway, and verify its robustness on synthetic data.
Schematic of gene expression models
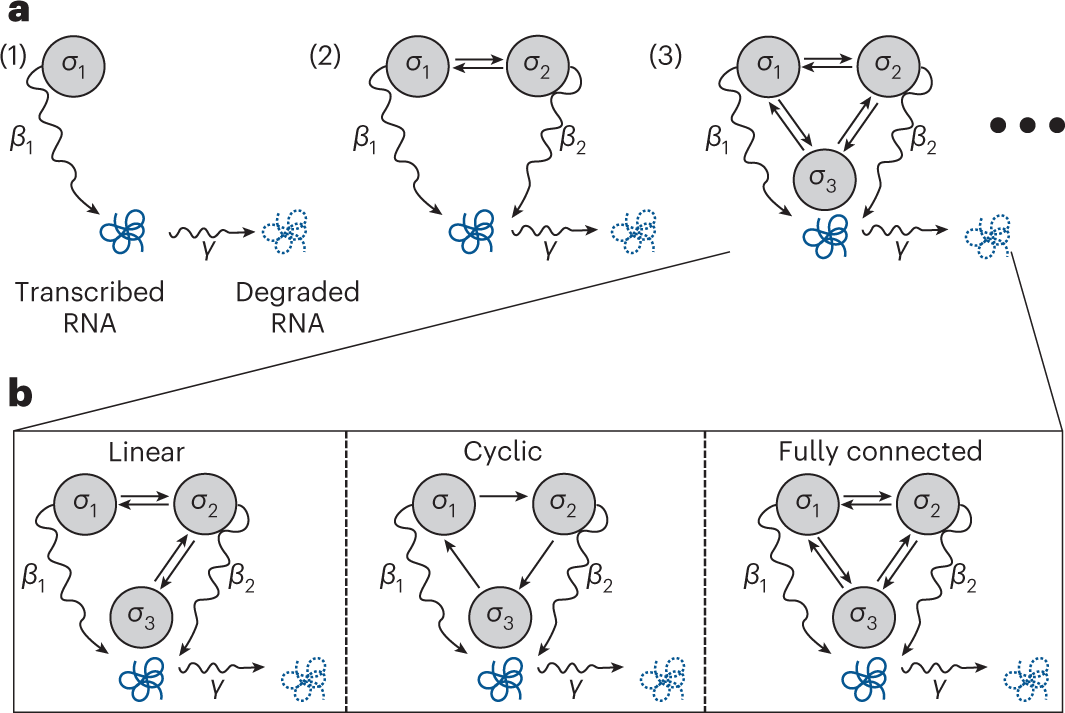
a, Schematic representations of one, two and three gene states. Each gray circle depicts an RNA production state (σi) that a gene may occupy, differentiated by its unique production rate βi. Straight arrows reflect possible transitions between gene states, and curved arrows depict RNA transcription (with rate β) or degradation (with rate γ). b, Models with a variety of transitions omitted. It is possible to develop a method to infer gene expression models (which includes gene state numbers and associated rates) alongside ‘connectivities’ directly from the data.
Availability – https://zenodo.org/record/7425217#.Y9plJnbMKUk
Kilic Z, Schweiger M, Moyer C et al. (2023) Gene expression model inference from snapshot RNA data using Bayesian non-parametrics. Nat Comput Sci [Epub ahead of print]. [abstract]


